Decoding Virus–Peptide–Cell Interactions
Published in Chemistry
The inherent ability of viruses to infect cells and deliver foreign genes is not always harmful – in fact, it can also be exploited for medical applications. Gene therapy holds great promise in treating otherwise incurable diseases and heavily relies on the ability of biotechnologically modified viruses to enter cells. However, these therapeutic viruses need to be very infectious to maximize the therapeutic outcome, which is why additives are required to boost their infectivity. Certain peptides can facilitate the transport of these viruses into cells. In this work, we have unveiled the molecular and physicochemical determinants of their activity.
Where did we start?
The roots of this project go back to 2013, to a study of Yolamanova, Weil, Münch, and coworkers.[1] They investigated the HIV-1 glycoprotein envelope through high-throughput screening assays and discovered a small protein fragment called “EF-C”. This peptide spontaneously self-assembles into supramolecular fibers with quite interesting properties: The fibers can act as a “glue” between viruses and cells, facilitating the delivery and uptake of viruses into cells which strongly improves efficiency for medical applications. The reason why these peptide fibers were so active was primarily attributed to their positive charge, which helped overcome the natural electrostatic repulsion between viruses and cells. However, back then this question had not been studied in detail.
Small changes have large effects
Because the short 12-mer peptide EF-C is easy to make in the lab, we used this peptide as a starting point to explore the effect of sequence variations. In an initial approach, we aimed to optimize the sequence of the EF-C peptide. While we succeeded in reducing the length of the sequence while maintaining a high bioactivity, we were surprised to find that certain changes in the molecular structure had very strong effects while other’s did not.[2] Let’s look, for example, at the peptides CKIKIQI (active) and CKIKQII (inactive). Both peptides have a net positive charge and form fibers, but it seems that this is not the determining feature as originally hypothesized. This finding raised some important questions: What physicochemical factors are essential for enhancing viral infectivity? To what extent does the molecular sequence influence structure formation? And how do these aspects relate to each other?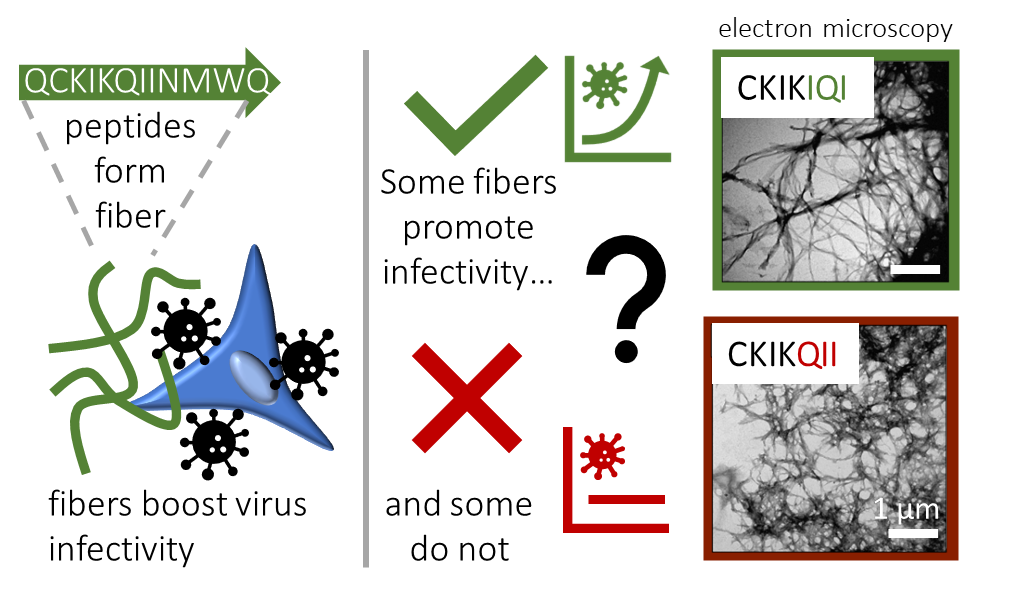
To answer these questions, we analyzed more than 150 different peptides that are derived from the original protein fragment EF-C.[3] We obtained this library of peptides by systematically replacing individual amino acids, truncating and extending the original sequence.
Multiparameter regression reveals interdependent properties
A challenge in establishing a structure–property–activity relationship for self-assembling peptides lies in the fact that the relevant properties span multiple length scales. Therefore, analysis methods that determine properties at various dimensions are required to investigate their intricate relationships systematically. We employed data-mining with regression models with the aim to simplify interdependent properties. This approach was chosen due to its ability to reveal underlying principles and multi-parameter patterns within a database.
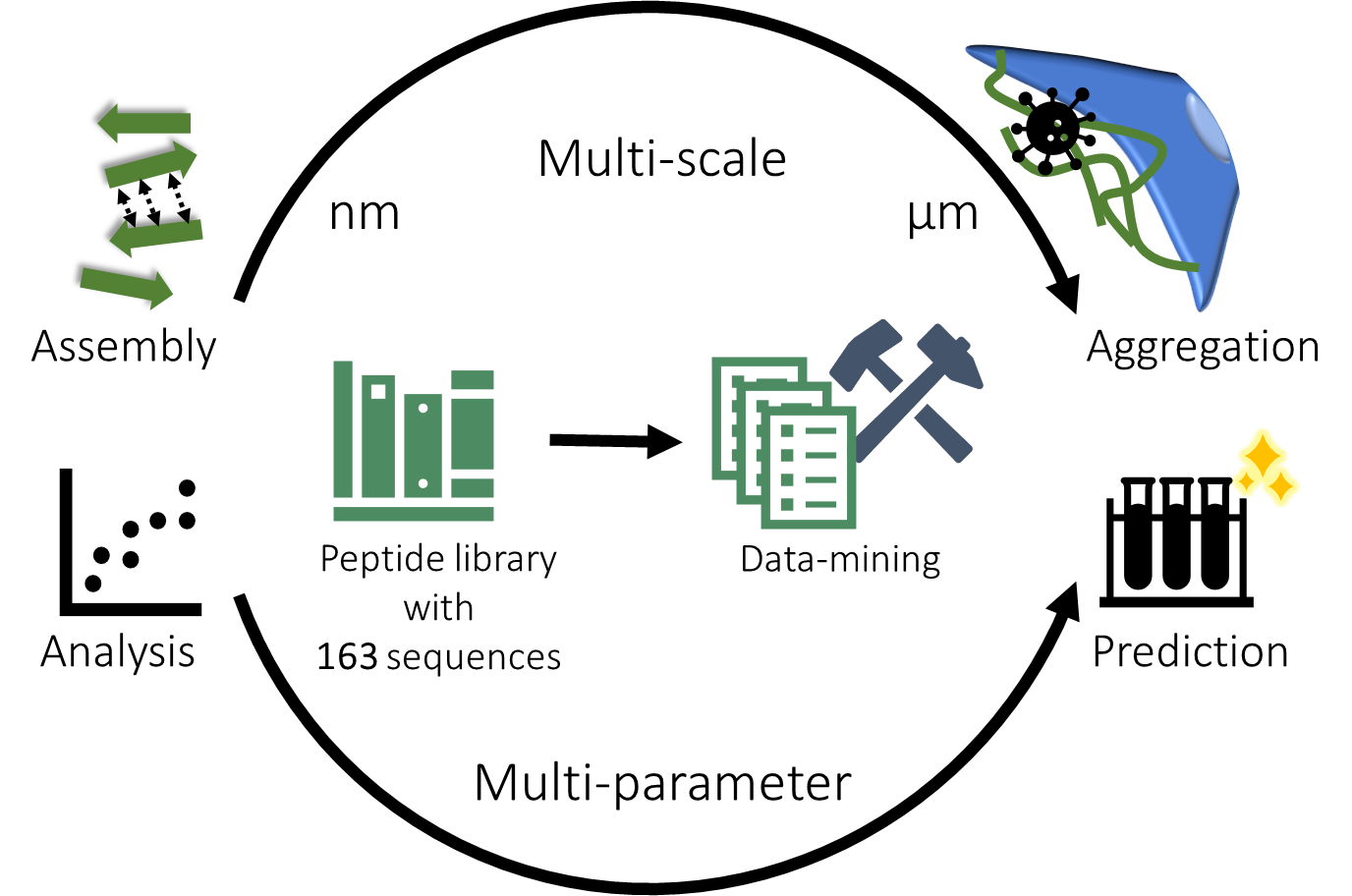
Viral infectivity is governed by hydrophobicity, charge and aggregation
In our data-mining analysis, we discovered that it is not the fibrils themselves but rather their µm-sized aggregates that enhance infectivity. Furthermore, peptides that boost viral infectivity exhibit hydrophobic structures rich in β-sheet content with a positive charge. Thus, the virus–peptide–cell interaction is not solely governed by fibril formation or electrostatic interactions, but also by interdependent features such as hydrophobicity and aggregation, which all collectively contribute to the bioactivity.
However, aggregation at the µm scale is not typically investigated as most studies focus on peptide self-assembly at the nm scale. Our study emphasizes the importance of aggregation processes for activity, a phenomenon observed across all peptides within our systematically constructed library, as well as among sequence-wise unrelated naturally occurring peptides from diverse backgrounds. The importance of aggregation can be illustrated with our earlier example: CKIKIQI can form micrometer-sized aggregates, while CKIKQII does not which marks the key difference between the two peptides.
Where does this leave us?
Our study demonstrates that data-mining provides valuable insights into the bioactivity of self-assembling peptides, even with relatively small datasets. It also enables the development of efficient retroviral transduction enhancers through rational design. Recently, we showcased this by predicting active sequences from our experimental dataset using machine learning.[4] Strikingly, these predicted peptides validated our previously identified property-activity relationship, even if they are structurally unrelated to the dataset. This suggests that we have captured a universal property-activity relationship for promoting viral infectivity.
An open question that is left is why certain amyloid fibrils aggregate to µm-sized clusters while others remain isolated. Due to experimental limitations to characterize fiber-fiber interactions, we currently employ molecular dynamics simulations to shed light on the molecular mechanisms of multi-fiber aggregation. We believe that the insights gained from studying peptide fibrils for viral infectivity can not only enhance the effectiveness of gene therapy in the future but also guide fundamental research questions for the origins of the structural diversity of amyloid fibrils.
References
[1] M. Yolamanova, C. Meier, A. K. Shaytan, V. Vas, C. W. Bertoncini, F. Arnold, O. Zirafi, S. M. Usmani, J. A. Müller, D. Sauter, C. Goffinet, D. Palesch, P. Walther, N. R. Roan, H. Geiger, O. Lunov, T. Simmet, J. Bohne, H. Schrezenmeier, K. Schwarz, L. Ständker, W.-G. Forssmann, X. Salvatella, P. G. Khalatur, A. R. Khokhlov, T. P. J. Knowles, T. Weil, F. Kirchhoff, J. Münch, Nat. Nanotechnol. 2013, 8, 130. https://doi.org/10.1038/nnano.2012.248.
[2] S. Sieste, T. Mack, E. Lump, M. Hayn, D. Schütz, A. Röcker, C. Meier, K. Kaygisiz, F. Kirchhoff, T. P. J. Knowles, F. S. Ruggeri, C. V. Synatschke, J. Münch, T. Weil, Adv. Funct. Mater. 2021, 31, 2009382. https://doi.org/10.1002/adfm.202009382.
[3] K. Kaygisiz, L. Rauch-Wirth, A. Dutta, X. Yu, Y. Nagata, T. Bereau, J. Münch, C. V. Synatschke, T. Weil, Nat. Commun., 2023, 14, 5121. https://doi.org/10.1038/s41467-023-40663-6.
[4] K. Kaygisiz, A. Dutta, L. Rauch-Wirth, C. V. Synatschke, J. Münch, T. Bereau, T. Weil, Biomater. Sci. 2023, 11, 5251. https://doi.org/10.1039/D3BM00412K.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in