FBXO7 ubiquitinates PRMT1 to suppress PHGDH arginine methylation, serine synthesis, and tumor growth in hepatocellular carcinoma
Published in Cancer
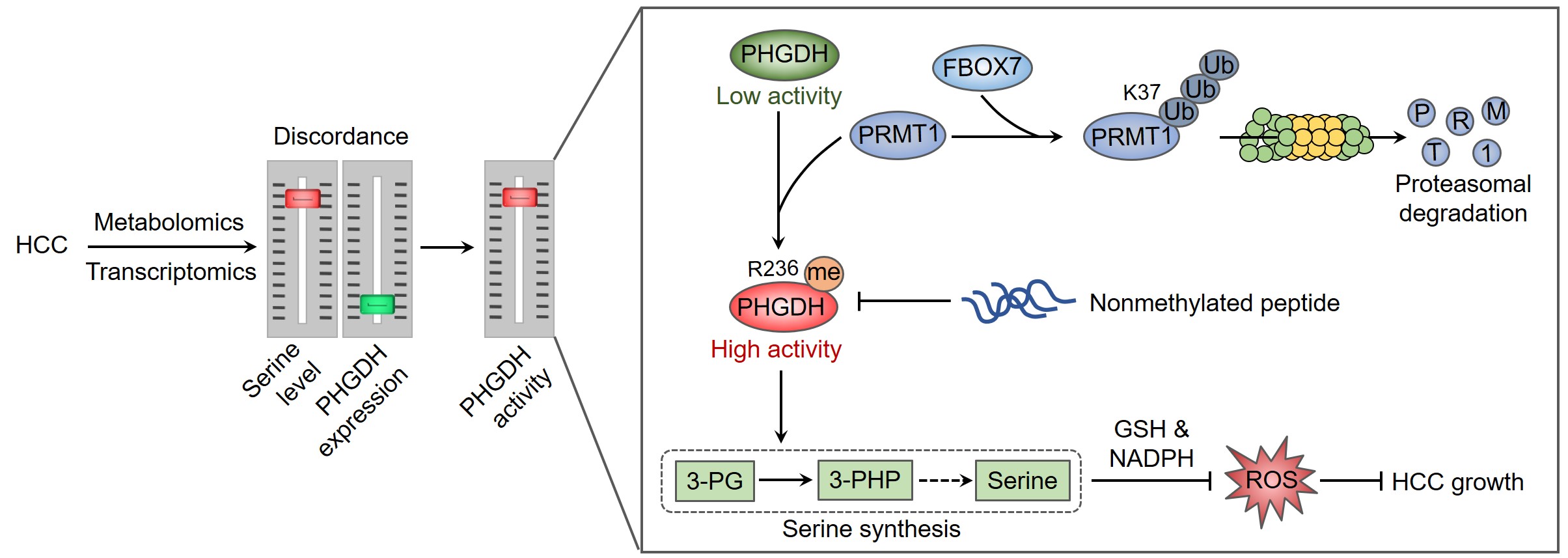
Nearly a century ago, metabolic changes were first observed in cancer cells by Otto Warburg, referring to as the Warburg effect. This work then laid the foundation for the continuously emerging findings regarding the mechanism and role of metabolic reprogramming in cancer onset, progression, and therapy. Among these metabolic abnormalities, the de novo serine synthesis, a side-branch of glycolysis, was found to hyperactivated in diverse cancer types. Starting with the conversion of 3-phosphoglycerate (3-PG, a glycolytic intermediate) to 3-phosphohydroxypyruvate (3-PHP) catalyzed by the rate-limiting enzyme phosphoglycerate dehydrogenase (PHGDH), serine can be generated through a three-step enzymatic reaction, and then converted to glycine to provide one carbon donor for one-carbon mechanism. Therefore, serine synthesis generates biomass and energy to support cancer cell proliferation. In addition, the intrinsic reductants (GSH and NADPH) can be produced in these processes to maintain redox balance of cancer cells 1, 2. Serine metabolism thus represents a favorable target for cancer therapy, but how serine synthesis is regulated in cancer remains largely unclear.
This story started with a previous report in our group to characterize the metabolic patterns of hepatocellular carcinoma (HCC) using HCC tissues and adjacent normal tissues 3, resulting from which an unexpected discordance between metabolomic and transcriptomic profiles in HCC was observed. Particularly, HCC tissues exhibited higher level of serine, but had significant lower levels of PHGDH mRNA and protein. Although this finding is in contrast to other reports that PHGDH is frequently overexpressed in cancer 4, 5, 6, it might be rationale for cancer cells to maintain a relatively low PHGDH level to endow metastatic potential 7. We then tried to explain how such a low level of PHGDH protein could support this large serine pool, and found that PHGDH was hyperactivated in HCC. The increase in PHGDH activity was attributable to protein arginine methyltransferase 1 (PRMT1)-mediated methylation of PHGDH at arginine 236. We have provided evidence demonstrating that PRMT1-mediated PHGDH methylation and activation potentiated serine synthesis, ameliorated oxidative stress, and promoted HCC growth both in cultured cells and animal tumor models. Using a site-specific antibody recognizing PHGDH arginine methylation, we found that PHGDH was hypermethylated at arginine 236 by PRMT1 in HCC tissues. Furthermore, we developed a nonmethylated therapeutic peptide to block PHGDH methylation, which exhibited remarkable anti-HCC effect in cooperation with serine and glycine restriction 3.
The hypermethylation of PHGDH requires high abundance of PRMT1 protein, but how PRMT1 is upregulated in HCC remain unclear. We presumed that the degradation of PRMT1 protein might be prevented, because the mRNA level of PRMT1 in HCC tissues was not obviously elevated compared with adjacent normal liver tissues. We thus employed an in silico screening combined with interactomics analysis to identify the interacting E3 ubiquitin ligases of PRMT1, pointing out F-box protein 7 (FBXO7) as a candidate. Further analysis has demonstrated a direct binding of FBXO7 with PRMT1, this binding led to PRMT1 ubiquitination at lysine 37 and promoted the proteosomal degradation of PRMT1. FBXO7-mediated PRMT1 degradation then prevented PHGDH methylation and activation, thereby inhibiting serine synthesis, aggravating oxidative stress, and suppressing HCC growth in cultured cells as well as animal tumor models. More importantly, we used HCC tumor samples to evaluate the clinical relevance of FBXO7-PRMT1-PHGDH axis, and found that FBXO7 was markedly downregulated in human HCC tissues, and inversely associated with the levels of PRMT1 protein and PHGDH methylation.
In summary, our study provides mechanistic insights into the regulation of serine metabolism by FBXO7-PRMT1-PHGDH axis in HCC, and suggests PHGDH methylation as a therapeutic vulnerability for HCC treatment. Of note, our findings imply that serine-targeting strategies using PHGDH inhibitors can be applicable for cancer patients with PHGDH hyperactivation, no matter whether PHGDH is overexpressed or not.
- Geeraerts SL, Heylen E, De Keersmaecker K, Kampen KR. The ins and outs of serine and glycine metabolism in cancer. Nat Metab 3, 131-141 (2021).
- Reina-Campos M, Diaz-Meco MT, Moscat J. The complexity of the serine glycine one-carbon pathway in cancer. J Cell Biol 219, (2020).
- Wang K, et al. PHGDH arginine methylation by PRMT1 promotes serine synthesis and represents a therapeutic vulnerability in hepatocellular carcinoma. Nat Commun 14, 1011 (2023).
- Locasale JW, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43, 869-874 (2011).
- Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346-350 (2011).
- DeNicola GM, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475-1481 (2015).
- Rossi M, et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature 605, 747-753 (2022).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in