
When I first joined the Engle lab as a graduate student in 2018, the group primarily focused on the directed functionalization of alkenes using palladium and nickel catalysts. While these methods had great synthetic value, I was interested in using the directing group as a tool for exploring the reactivity and coordination chemistry of early transition metals, which were less well-known for catalyzing alkene functionalization. I thought the directing group approach may allow us to identify previously overlooked reactivity and develop alkene functionalization reactions that were outside the scope of late transition metals.
Around the end of my first year, I attended a departmental seminar by Professor Naoto Chatani (Osaka University) and was inspired to try some isomerization–carbonylation reactions of alkenes using ruthenium and rhodium catalysts in combination with a directing group. In particular, Keary and I thought that Chatani’s NHPic amide directing group (Pic = 2-picolinyl), which had previously been used for C–H activation reactions,1 may be able to promote a tandem alkene isomerization–hydrofunctionalization due its high conformational flexibility.

Although this idea looked good on paper, I was surprised to find that there were seemingly no reports at the time on the use of directing groups to control tandem isomerization and functionalization of alkenes to achieve internal site-selectivity. I thought the directing group strategy could potentially enable a new mode of site-selectivity control, and due to the industrial importance of tandem alkene isomerization–carbonylation reactions (e.g., the oxo-process), I thought that this could be a worthwhile endeavor. After trying countless reactions with various directing groups and well known carbonylation catalysts based on rhodium, ruthenium, and cobalt, I was unable to obtain more than 10% of my desired product. I then thought that the strain required for some of the key elementary steps to occur must be too high for the coordination geometries typically adopted by these metals, and therefore we would likely need a new strategy.
At this point we came across interesting paper by Professor Harry Gray from 1974 in which he showed that simple W(CO)6 could isomerize alkenes through a W(0)/W(II) redox cycle.2 Additionally, I found a report on the use of bidentate directing groups to promote oxidative addition and reductive elimination in the W(0)/W(II) redox couple.3 These directing groups looked similar to the ones our lab has utilized for alkene functionalization, and in this case, they were able to isolate and characterize various 7-coordinate W(II) species, which showed very interesting changes in coordination geometry compared with octahedral W(0) species. The ability of the metal to adopt various 7-coordinate geometries seemed promising because it would allow the substrate and flexible directing group to adopt the ideal conformation needed to promote the various steps of the envisioned tandem catalytic process.
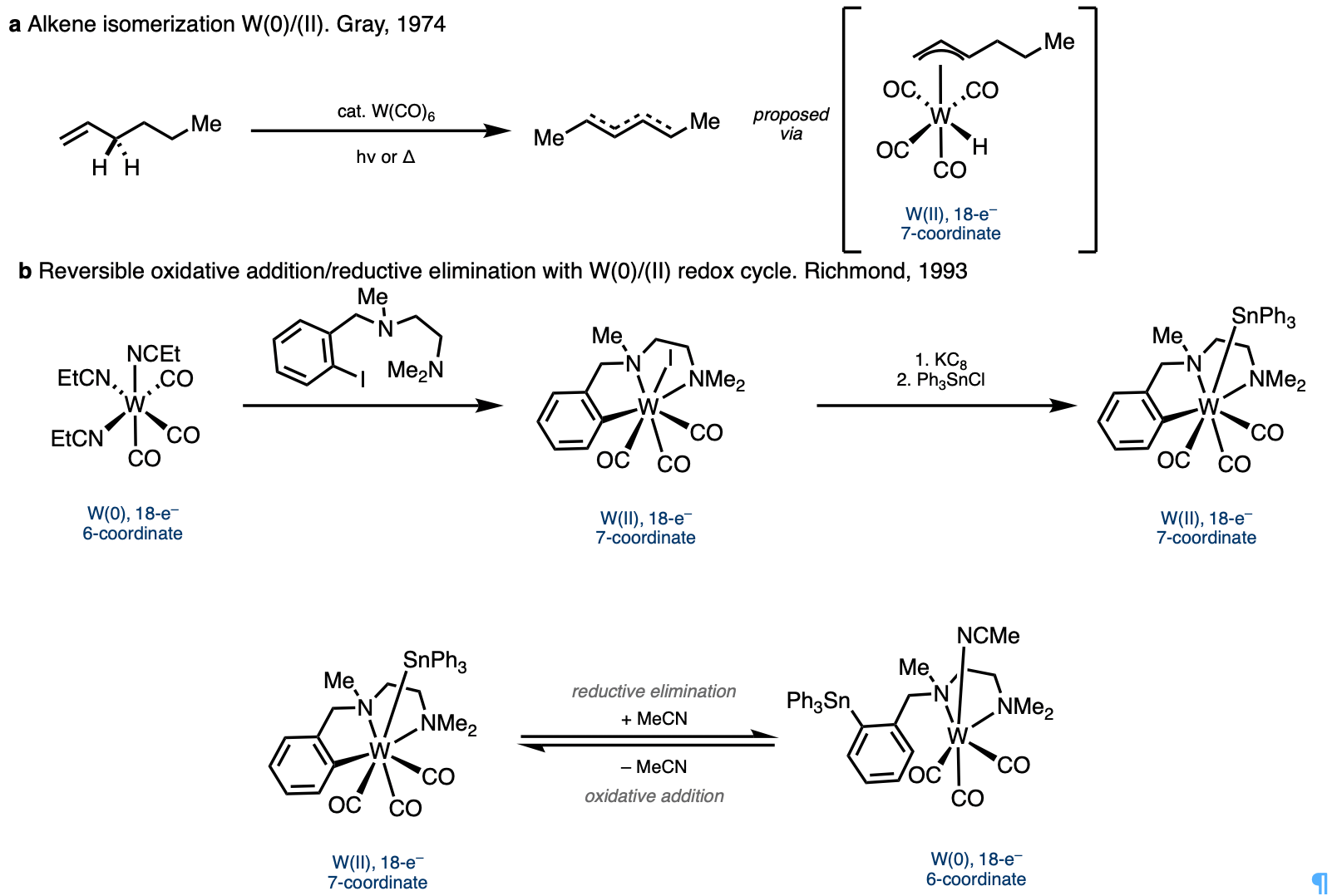
We began exploring the use of low-valent group-6 metals, and an ambitious visiting undergraduate student, Zi-Yang (Nick) Qin, joined me on this project and ran the first reaction with one equivalent of W(CO)6 using Chatani’s NHPic directing group appended to a distal alkene. To our amazement, he was able to isolate the product in over 50% yield! He then quickly synthesized substrates with alkenes at various distances from the directing group as well as some cyclic alkenes and tested them in the reaction the next day. After confirming the substrate generality of the reaction, Zi-Yang left to finish his undergraduate degree at USTC in Hefei, China.
After completing the substrate scope, I recognized that this study would be more beneficial to the chemical community if we were able to fully understand how this unique catalytic process was operating through rigorous mechanistic studies. After considering all possibilities, ranging from bimetallic processes to mechanisms involving Fischer carbenes, I thought the most reasonable possibility was a W(0)/W(II) redox cycle where N–H oxidative addition and C–N reductive elimination were occurring.
I initiated mechanistic work by adding my starting material to the W(MeCN)3(CO)3 pre-catalyst at room temperature to potentially detect an intermediate by in situ 1H NMR. However, after 10 minutes I could only observe starting material and product, with no detectable intermediates. At this point I realized that other approaches to studying the mechanism would be needed, and we teamed up with our computational collaborators, Prof. Peng Liu and his group at University of Pittsburgh, and they eagerly took to the task of exploring a metal that was completely new to them involving a largely uncharacterized mechanism.
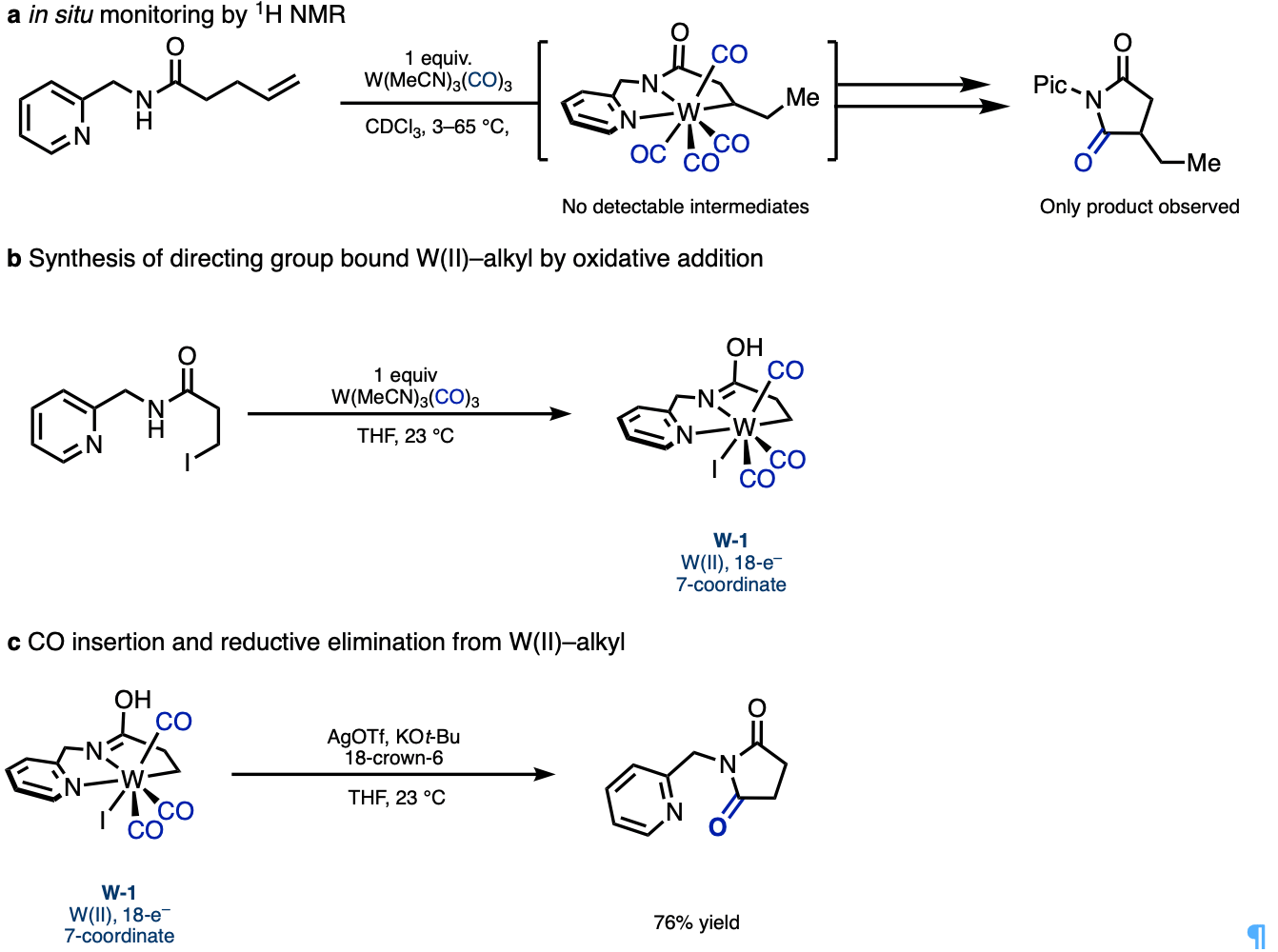
While they were hard at work investigating potential pathways computationally, I pursued the direct synthesis of a directing-group-bound W(II)–alkyl species. As these putative intermediates could not be directly observed by hydride insertion to the alkene starting material in the NMR experiments described above, we would need to find a completely different synthetic route. Alkyl–I oxidative addition with W(0) was unknown at the time, but I thought if it was possible, this would be best way to synthesize a directing group bound W(II)–alkyl species. The resulting iodide ligand from after the oxidative addition, would render the tungsten center more electron-rich, and hopefully suppress CO migratory insertion and reductive elimination to facilitate isolation of the desired W(II)–alkyl model complex.

After successfully preparing this model complex, CO migratory insertion and reductive elimination could then be promoted thermally or by the addition of silver to remove the iodide and base to absorb the HI created from reductive elimination. Our computational collaborators were able to support this pathway and elucidate many of the different 7-coordinate intermediates and transition states. With strong experimental and computational evidence for many of the key intermediates and elucidation of the unique coordination chemistry of W(II), we felt confident to share this story.
After submitting to Nature Chemistry, we were delighted to receive the reviewers’ comments, who were supportive of publishing our work. While this work was under review, I was also able to apply the mechanistic insights from this work to guide a new study on a W(0)-catalyzed tandem alkene isomerization–hydroboration reaction4 that addresses some of the limitations of the present study, such as the need for high temperature, a stoichiometric auxiliary, and relatively high catalyst loading. I hope this work in Nature Chemistry will revitalize interest in low-valent tungsten as a catalyst for alkene functionalization, and that our findings will provide insights to the unique coordination chemistry and reactivity of the W(0)/W(II) redox couple.
References
- Inoue, S.; Shiota, H.; Fukumoto, Y.; Chatani, N., Ruthenium-Catalyzed Carbonylation at Ortho C−H Bonds in Aromatic Amides Leading to Phthalimides: C−H Bond Activation Utilizing a Bidentate System. Journal of the American Chemical Society 2009, 131 (20), 6898-6899.
- Wrighton, M.; Hammond, G. S.; Gray, H. B., Group VI metal carbonyl photoassisted isomerization of olefins. Journal of Organometallic Chemistry 1974, 70 (2), 283-301.
- Buffin, B. P.; Poss, M. J.; Arif, A. M.; Richmond, T. G., Synthesis and reactivity of a tungsten(0) anion stabilized by chelating tertiary amines. The oxidative addition and reductive elimination of a carbon-tin bond at tungsten. Inorganic Chemistry 1993, 32 (18), 3805-3806.
- Jankins, T. C.; Martin-Montero, R.; Cooper, P.; Martin, R.; Engle, K. M., Low-Valent Tungsten Catalysis Enables Site-Selective Isomerization–Hydroboration of Unactivated Alkenes. Journal of the American Chemical Society 2021, 143 (37), 14981-14986.
Follow the Topic
-
Nature Chemistry
A monthly journal dedicated to publishing high-quality papers that describe the most significant and cutting-edge research in all areas of chemistry, reflecting the traditional core subjects of analytical, inorganic, organic and physical chemistry.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in