
Single-cell and spatial molecular profiling technologies enable scientists to obtain individual gene expression profiles for large numbers of single cells, including cells isolated from patients, allowing us to interrogate the cellular and molecular basis of biological systems in a way previously unimagined. These new technologies let us profile tumors, embryos, tissues as they truly are: dynamic, multicellular, spatially-organized systems that evolve and develop over time. Briefly, single-cell and spatial molecular technologies profile individual cells without population-level homogenization. This captures the state of a system at a time point, giving us richer information about within-type variation, and tells us about cell types but goes beyond marker genes to include readouts of cellular function. Indeed, the combination of cellular annotations and cellular phenotypes may often matter immensely to the biology of the system. Cell fate decisions and disease states result from complex spatiotemporal dynamics spanning molecular, cellular, tissue, and environmental factors. This leaves the bioinformatics field at large questioning how to computationally distinguish cell type from cell state and, ultimately, cells from each other based on large-scale datasets describing their molecular profiles.
Single-cell transcriptomics datasets typically take the form of a matrix with one dimension of at least 20 thousand genes and the other dimension ranging from several thousand to several hundred thousand cells. The size of these datasets are continually growing in both dimensions as technology develops to refine multi-omics characterization of cells at scale. This high dimensional nature challenges our goal of distilling only a few readouts of cellular behavior and features of interest for interpretation. Single-cell analysis differs from sorting and studying cells using flow cytometry, where the readouts are few and carefully chosen to stain the surfaces of specific cell populations. With tens of thousands of molecular readouts, however, we get information about many cell types at once but are faced with the problem of not knowing where to look in the dataset. The goal of single-cell data analysis is to refine and distill this molecular data into interpretable statistics, so we need tools that let you infer which markers are most relevant to cellular function in a data-driven, unbiased way. The unbiased, comprehensive nature of single-cell experiments is our greatest strength for deriving new biological knowledge and mechanistic insights into biological systems.
Although single-cell assays are high dimensional, we hypothesize a smaller number of relevant cellular phenotypes. This concept is commonly employed in cell type identification through clustering. For example, in white blood cell single-cell sequencing data, we are consistently able to split which cells came from myeloid vs lymphoid lineages as real lineage differences manifest in clear, consistent bifurcation of two groups. This provides a convenient method for sorting cells into groups based on the most variable features in the data matrix. While powerful, clustering limits cells to single labels and identities. A cell may not simultaneously belong to cluster X and Y, which becomes problematic in certain situations. Especially in immunology, gene expression is stochastic and populations often comprise cells reacting within a spectrum rather than an army of identical soldiers. For example, if we imagine cluster X is labeled as “CD4 T cells” and Y as “tissue-resident T cells”. By forcing these groups apart, we may be missing important statistics about their similarities. We want to discover gene signatures that are associated broadly with programs of cell behavior. Increasingly we are understanding some cell state transitions as continuous processes rather than discrete stages of development. A notable example would be M1 and M2 macrophages, which are now increasingly understood to be two polarization states rather than two differentiated types.
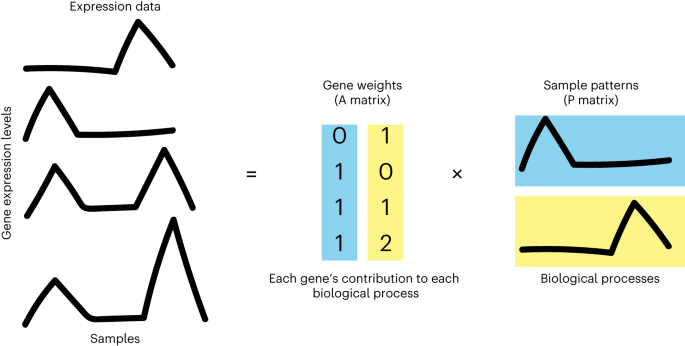
There is a need for methods to obtain continuous gene signatures to define continuous patterns of cellular function across a large number of sample conditions and measured features. This problem has already been solved for other types of large data and for bulk gene expression1. Matrix factorization is a type of machine learning that is used to find patterns in large datasets. Non-negative matrix factorization (NMF) is a version of this specifically optimized for non-negative data2–4, and has a long history of application to transcriptional datasets5. Data types well suited for NMF include audio signal, astronomy data, netflix movie ratings, and gene counts--it has been said there are no negative quantities in nature6. The biological concept of single-cell RNA counts data means that the interpretation of NMF is used specifically for finding patterns of gene expression (representing cell function, state, response) in a large group of cell measurements. Briefly, NMF is a method that decomposes/factorizes input data into two matrices whose product approximates the input data. One of the matrices, called the Amplitude matrix, is of dimension number of genes n by number of patterns p, and the other, called the Pattern matrix, is of dimension number of patterns p and experimental conditions m. The two matrices complement each other; together, they describe the association between patterns and genes (Amplitude matrix) and associations between those same patterns and each condition in the dataset (Pattern matrix). Our group extended a Bayesian NMF method called CoGAPS7 to specific features to account for size and sparsity of single-cell data8 and have continued to extend features and vignettes towards interpretation of scRNA-seq data features biologically as demonstrated in our Nature Protocols publication.
Once the algorithm solves for the amplitude and pattern matrices, how do you actually learn and interpret biology from them? In our publication we describe methods to associate patterns with annotated sample groups and to associate patterns with gene sets curated to reflect currently theorized involvement in well-studied biological processes. The ultimate translational goal of this analysis is to identify new gene candidates associated with cellular disease processes to investigate at the bench. There are few available software packages specific to biological NMF and few workflows to perform biological interpretation of the resulting patterns. Our goal is interpretation of NMF solutions in a biologically meaningful way with these packages--therefore, protocol is organized around: (1) How to identify the right number of patterns for your biological question of interest to uncover the right “level” of variation for your scientific question? (2) How to define marker genes of cellular phenotypes from the amplitude matrix? (3) How to determine the likely function of these groups of marker genes? (4) How to interpret cellular features and phenotypes that distinguish annotated conditions in the dataset? This allows you to quickly assess whether patterns are “capturing” the broad cell groups or processes currently hypothesized to exist in your single-cell dataset. To provide guidelines to support this analysis, our protocol presents a full suite of tools which let you run CoGAPS on single-cell data, perform post-hoc statistics and visualization of NMF results, and grants the flexibility to use R, Python, GenePattern Notebook, or a Docker deployment. We demonstrate these features to explain how CoGAPS works in the context of a cohort of single-cell RNA-seq data from pancreatic cancer.9 This workflow can be applied directly to other datasets, or can be adapted to extend the same functions to alternative implementations of other models for NMF and to other continuous latent space analysis methods of single-cell data.
Looking forward to the next decade, we anticipate new data types, new data formats, and new types of data altogether. Most recently, single-cell dissociation assays have given way to new spatial molecular technologies. Spatial data can be analyzed with the same process10. As new forms of data emerge, one strategy for tackling them could be to adopt tools from other areas of science (with similar data types and dimensionality problems) and apply them to the data from these new assays to learn new biology. Multi-omics data also can be extended in matrix factorization either through coupled MF or transfer learning.11,12 In the multi-omics field, some researchers may want to apply multiple high-dimensional assays to capture all of the information necessary for their experiment12. Once you have results for each, an important area of research is how to connect them13. While powerful, we note that NMF is one of many dimensionality reduction tools and linear methods may have challenges for some types of cellular behaviors. To complement technology development, the research community is also undergoing a revolution in the rapid advances to artificial intelligence methods. Many unsupervised learning methods are emerging in the artificial intelligence era, but the focus on methods to ensure biological interpretability described in this proposal will remain important when applying these methods to infer relevant cellular features from high dimensional datasets.
References
- Ochs, M. F. et al. Detection of Treatment-Induced Changes in Signaling Pathways in Gastrointestinal Stromal Tumors Using Transcriptomic Data. Cancer Res. 69, 9125–9132 (2009).
- Ochs, M. F., Stoyanova, R. S., Arias-Mendoza, F. & Brown, T. R. A New Method for Spectral Decomposition Using a Bilinear Bayesian Approach. J. Magn. Reson. 137, 161–176 (1999).
- Wang, G., Kossenkov, A. V. & Ochs, M. F. LS-NMF: A modified non-negative matrix factorization algorithm utilizing uncertainty estimates. BMC Bioinformatics 7, 175 (2006).
- Lee, D. D. & Seung, H. S. Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791 (1999).
- Brunet, J.-P., Tamayo, P., Golub, T. R. & Mesirov, J. P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. 101, 4164–4169 (2004).
- Stein-O’Brien, G. L. et al. Enter the Matrix: Factorization Uncovers Knowledge from Omics. Trends Genet. 34, 790–805 (2018).
- Fertig, E. J., Ding, J., Favorov, A. V., Parmigiani, G. & Ochs, M. F. CoGAPS: an R/C++ package to identify patterns and biological process activity in transcriptomic data. Bioinformatics 26, 2792–2793 (2010).
- Sherman, T. D., Gao, T. & Fertig, E. J. CoGAPS 3: Bayesian non-negative matrix factorization for single-cell analysis with asynchronous updates and sparse data structures. BMC Bioinformatics 21, 453 (2020).
- Kinny-Köster, B. et al. Inflammatory Signaling in Pancreatic Cancer Transfers Between a Single-Cell RNA Sequencing Atlas and Co-Culture. http://biorxiv.org/lookup/doi/10.1101/2022.07.14.500096 (2022) doi:10.1101/2022.07.14.500096.
- Deshpande, A. et al. Uncovering the spatial landscape of molecular interactions within the tumor microenvironment through latent spaces. Cell Syst. 14, 285-301.e4 (2023).
- Liu, J. et al. Jointly defining cell types from multiple single-cell datasets using LIGER. Nat. Protoc. 15, 3632–3662 (2020).
- Lê Cao, K.-A. et al. Community-wide hackathons to identify central themes in single-cell multi-omics. Genome Biol. 22, 220 (2021).
- Sharma, G., Colantuoni, C., Goff, L. A., Fertig, E. J. & Stein-O’Brien, G. projectR: an R/Bioconductor package for transfer learning via PCA, NMF, correlation and clustering. Bioinformatics 36, 3592–3593 (2020).
Follow the Topic
-
Nature Protocols
This journal publishes secondary research articles and covers new techniques and technologies, as well as established methods, used in all fields of the biological, chemical and clinical sciences.






Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in