Mutation in Bruton Tyrosine Kinase (BTK) A428D Confers Resistance To BTK-Degrader Therapy In Chronic Lymphocytic Leukemia
Published in Cancer, Genetics & Genomics, and Pharmacy & Pharmacology
Targeting Bruton's Tyrosine Kinase (BTK) has profoundly changed the face of treatment for patients with chronic lymphocytic leukemia (CLL). Iterative advances in the cat and mouse game of resistance and redesign have moved BTK inhibitors from covalent to non-covalent and now targeted protein degraders.
In the journal Leukemia 38:1818 (2024), Kipps and colleagues describe a patient who developed successive resistance to a covalent BTKi, then a non-covalent BTKi, and then a BTK-degrader through acquisition of distinctive mutations in BTK. Each mutation in BTK depicted in red font in the figure conferred resistance to the ongoing BTKi therapy, as indicated on the top of the figure.
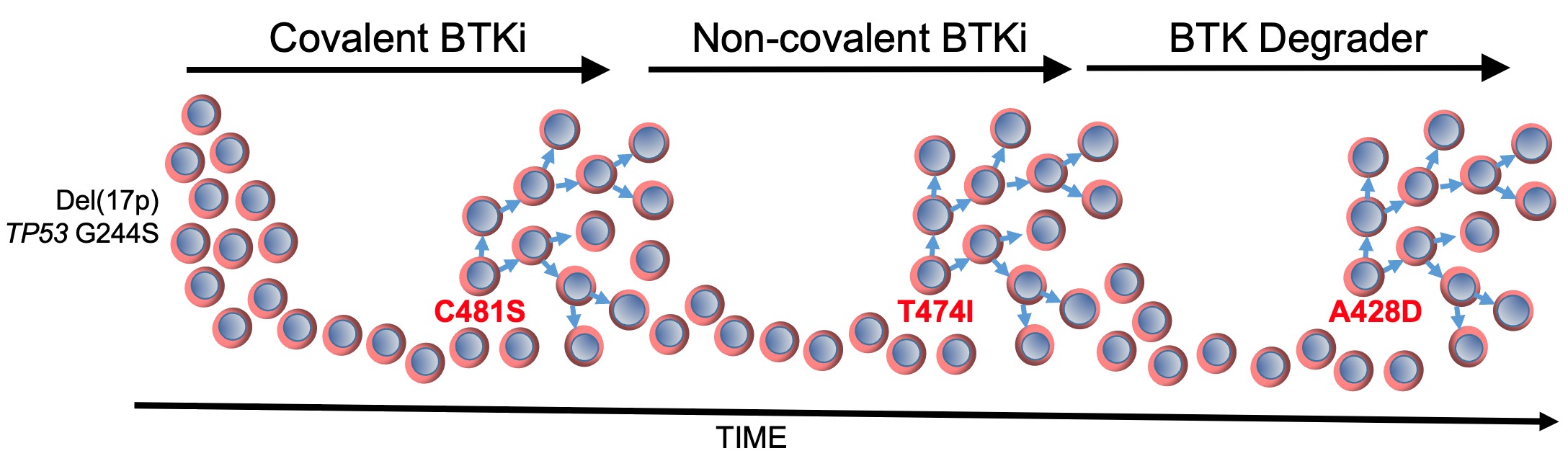
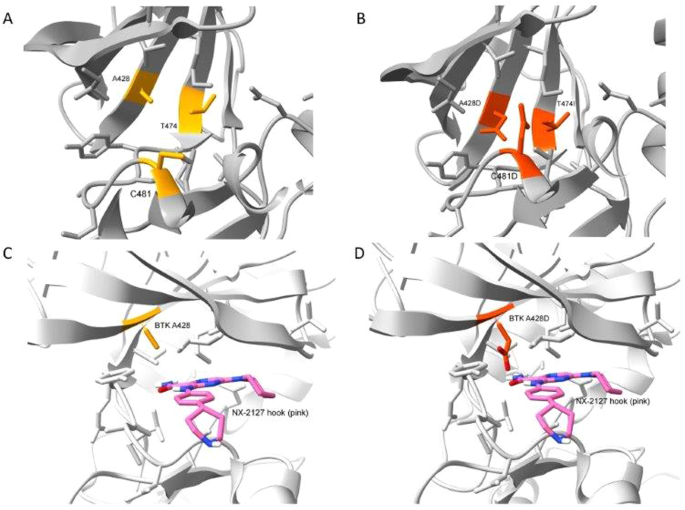
The first BTK mutation to develop was C481S, which was found to block the capacity of covalent BTK inhibitors, such as ibrutinib, acalabrutinib, or zanubrutinib, to form a chemical bound with BTK (Woyach JA, J Clin Oncol 35:1437, 2017), thereby reducing their capacity to block BTK and B-cell receptor signaling. The second BTK mutation to develop was T474I, which has been found to impair the clinical activity of non-covalent BTK inhibitors, such pirtobrutinib, and cause resistance to therapy with covalent BTK inhibitors (Wang E, et al, N Engl J Med 386:735, 2022). Finally, the third BTK mutation was A428D, which had been observed in earlier studies as being associated with resistance to both covalent and non-covalent BTK inhibitors (ibid). Like BTK mutation L528W, the BTK mutation at A428D substantially reduces its enzymatic activity, impairing its capacity to effect autophosphorylation at Y223 and phosphorylation of PLCγ2 at Y1217; however, these so-called “dead kinases” retain their capacity to induce downstream BTK signaling through non-enzymatic means to effect activation of AKT and ERK, like wild-type BTK. However, unlike L528W, the BTK mutation A428D introduces an aspartic acid in place of an alanine at a site in the kinase domain that appears to be recognized by the “hook” attached to an E3 ligase, Cereblon, which when bound to BTK causes its polyubiquitination and subsequent proteasomal degradation. The steric interference caused by this amino acid substitution apparently mitigates the binding of the BTK-degrader to BTK, thereby resulting in drug resistance. Each of these BTK mutations appeared in isolation of the others, suggesting that a BTK carrying more than one BTK mutation may be unable to mediate effective downstream signaling required for disease progression and clinical resistance to therapy.
Despite observing disparate subclones with each successful therapy failure, each of these subclones apparently originated from the same base ‘mother clone’ of CLL that harbored del(17p), mutation in TP53 (G244S), and complex karyotype. The TP53 mutation (G244S) observed in the CLL cells of this patient encodes a mutant TP53 protein that has been identified in a family with Li Fraumeni syndrome, in which the inheritance of this mutant TP53 confers an extremely high risk of developing cancer (Hu, H, et al, Sci Rep 6:20221, 2016). This mutant TP53 lacks the wildtype TP53 function of being able to induce expression of CDKN1A, which encodes p21 (ibid). The protein p21 plays a essential role in regulating the cell cycle by inhibiting cyclin-CDK complexes, thereby causing cell cycle arrest at the G1 phase. The inability to induce p21 by this mutant TP53 can allow for unchecked proliferation of each mutant subclone when it becomes immune from the ongoing BTKi therapy, resulting in disease progression on each type of BTK inhibitor, including a BTK-degrader.
This case illustrates that a mutation in the kinase domain of BTK, namely A428D, can confer disease resistance to a BTK degrader, namely BGB-16673. Modeling of a BTK A428D mutation places a negatively charged aspartic acid in place of the hydrophobic side chain of alanine within the binding pocket of another BTK-degrader also in clinical development, namely NX-2127, suggesting that CLL cells with BTK A428D also may be resistant to NX-2127, as they already are known to be with either non-covalent or covalent inhibitors of BTK. Consequently, the two BTK degraders furthest advanced in clinical trials potentially may select for CLL cells with BTK A428D that are resistant to all approved BTKi’s.
Follow the Topic
-
Leukemia
This journal publishes high quality, peer reviewed research that covers all aspects of the research and treatment of leukemia and allied diseases. Topics of interest include oncogenes, growth factors, stem cells, leukemia genomics, cell cycle, signal transduction and molecular targets for therapy.





Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in