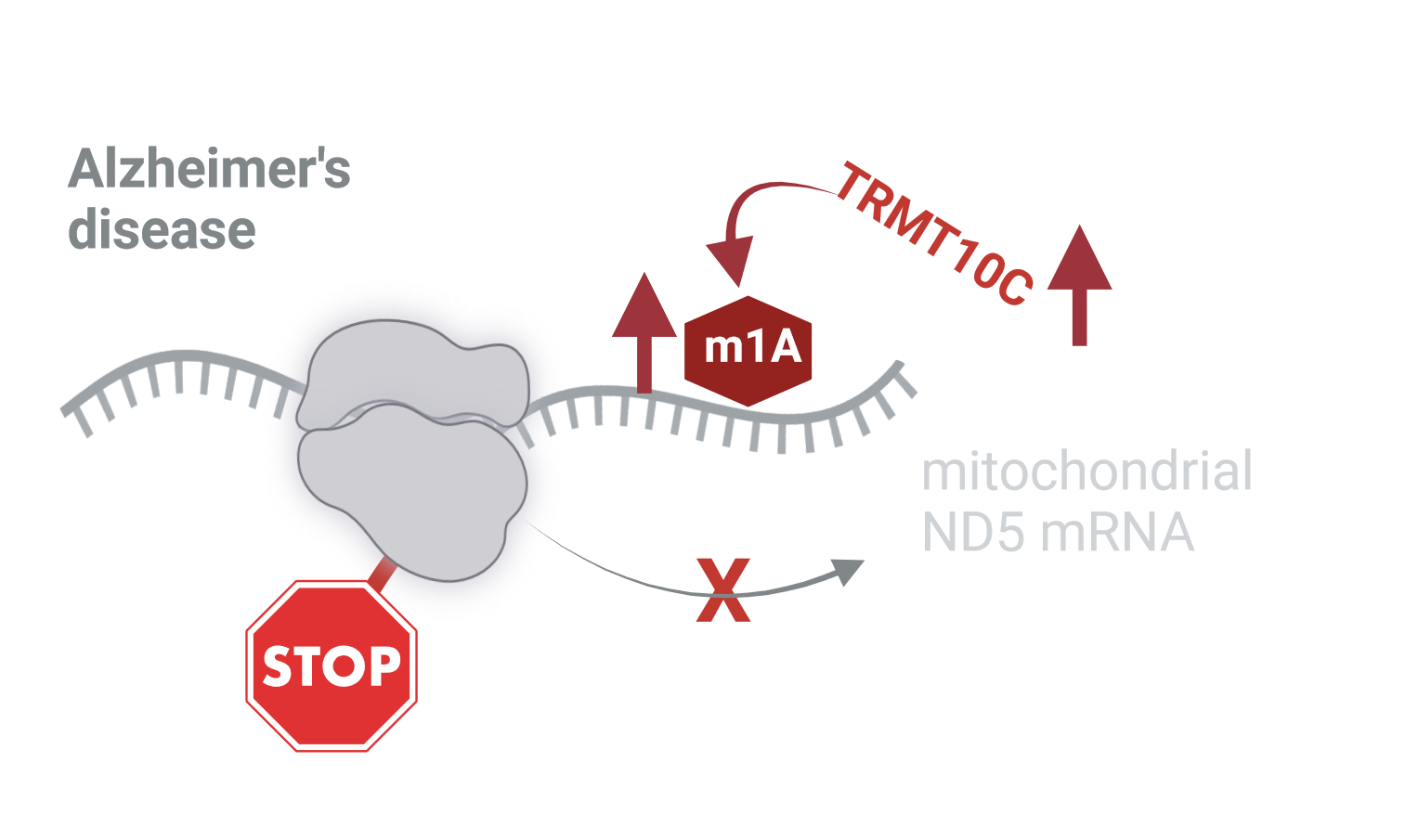
N1-methylation of adenosine (m1A) in ND5 mRNA leads to complex I dysfunction in Alzheimer’s disease
Published in Chemistry, Neuroscience, and Cell & Molecular Biology

![]() What we already knew:
What we already knew:
Mitochondrial dysfunction is one of the major pathologic hallmarks in sporadic Alzheimer's disease (AD), the most common form of AD with ageing as one important risk factor. Several alterations in mitochondrial function were associated with AD strongly associated with reduced effectivity of the respiratory chain especially complex I comprising impairment of complex I activity and reduced protein and mRNA levels of its nuclear-encoded subunits in multiple zones of post-mortem AD brain. Data dealing with mitochondrial-encoded subunits of complex I such as ND5 are still missing. ND5 is one of 7 mitochondrial encoded subunits and is located at the most distant position of the membrane arm. It provides a long transversal helix aligned parallel to the membrane arm which transmits the energy released by the redox reaction in the peripheral arm to proton translocation in the membrane arm. Therefore, ND5 is considered essential for the coupling of oxidation and proton transport. In recent AD genome-wide association studies, no single nucleotide polymorphisms in genes coding for complex I were identified, pointing to epigenetic or posttranscriptional mechanisms regulating complex I activity and expression.
One important but until now underexplored post-transcriptional mechanism to determine accuracy and optimal rate of translation in AD are RNA modifications. More than 170 different types of these alterations, which are thought to fine-tune RNA function have been identified. They abundantly occur on transfer RNA and ribosomal RNA. In contrast, occurrence, quantity and positioning of mRNA modification, especially methylation, are still highly discussed. One of these controversial mRNA methylations is m1A. One m1A modification coherently determined is the mitochondrial ND5 mRNA m1A at position 1374. Biosynthesis of m1A is catalyzed by the writer methyltransferase TRMT10C. Because of the location of the methylgroup in m1A on the Watson-Crick face, ND5 mRNA methylation on position 1374 within codon GCA is thought to disrupt base pairing during decoding on the mitoribosome, thereby impeding effective translation of ND5.
What was missing:
Until now, there is no direct experimental evidence that ND5 m1A mRNA methylation leads to reduced mitochondrial function or might even be involved in pathological alterations of mitochondrial function.
Therefore, we asked three major research questions in this publication:
(1) Are the protein levels of the m1A writer enzyme TRMT10C altered in AD cell model as well as in AD patients?
(2) Is m1A methylation of ND5 mRNA consequently increased in AD?
(3) Does this lead to reduced ND5 protein expression and via this causal chain to impaired mitochondrial function?
What we did and what we discovered:
We adopted a multidisciplinary approach using an AD cell model, human AD post mortem brain tissue as well as publicly available RNAseq databases from AD patients. Our AD cell model is a HEK293 cell model overexpressing stably overexpressing the wild-type human Amyloid precursor protein (APPwt), which displays a 10-fold increased Aβ1-40 level. The post-mortem frontal cortex samples (Gyrus frontalis superior 3+4) from AD patients and aged matched controls were received from The Netherlands Brain Bank. RNA-Seq data from two human databases were analyzed. The first was data from Aging, Dementia and Traumatic Brain Injury study, in which the transcriptome of four different brain regions (White matter of Parietal Cortex, Hippocampus, Parietal Cortex, Temporal Cortex) was analyzed. Furthermore, we used single-cell transcriptomic analysis examining 6 major brain cell types from the frontal cortex of 24 AD patients and 24 age-matched controls (Mathys et al. 2019 Nature).
Using this broad set of models and methods, we provide first evidence that TRMT10C protein levels are increased in our AD cell model, in post-mortem tissue and RNA-Seq data from AD patients, plausibly leading to increased m1A methylation of ND5 mRNA. Importantly, TRMT10C mRNA levels are only elevated in inhibitory and excitatory neurons already at an early stage of the disease. Increased levels of methylated ND5 mRNA correlate with reduced ND5 protein levels in AD models and patients. Similarly, we demonstrate that TRMT10C overexpression in HEK293 cells results in increased m1A methylation of ND5 and reduced ND5 protein levels consequently leading to reduced mitochondrial function reflected in reduced respiration and mitochondrial membrane potential. This newly described mechanism driving mitochondrial dysfunction in AD is both driven by oligomeric Aß1-42 as well as oxidative stress. In addition, we are able to rescue ND5 protein expression HEK APPwt cells knocking down TRMT10C using siRNA, which also reduces m1A levels in these cells.
Our conclusion
Together, these results prompt us to conclude that m1A methylation of ND5 mRNA might fine-tune mitochondrial function under physiological conditions to the energetic needs of the cells. Under pathophysiological conjectures e.g. of enhanced mitochondrially generated ROS or elevated Aβ levels crossing the threshold of healthy aging toward pathological aging and AD, TRMT10C protein expression and m1A methylation levels of ND5 mRNA are strongly elevated resulting in repression of ND5 protein translation. Together with the Aβ induced inhibition of protein transport into mitochondria, reduced mRNA expression as well as reduced protein expression of several nuclear encoded subunits of complex I, this newly described mechanism might be involved in the induction of mitochondrial dysfunction in AD and thereby represent an exciting new pathway which could be targeted for AD therapy.
Follow the Topic
-
Molecular Psychiatry
This journal publishes work aimed at elucidating biological mechanisms underlying psychiatric disorders and their treatment, with emphasis on studies at the interface of pre-clinical and clinical research.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in