Read2Tree infers phylogenetic trees from raw sequencing reads quick and easy
Published in Ecology & Evolution

In biology, phylogenetic trees are everywhere. They help us understand the relationships between species, genes, or cells—how they evolved, and how they're related.
The sequencing revolution provides the raw material to infer phylogenetic trees, but building state-of-the-art phylogenetic trees requires tedious steps from read curation, de novo assembly, gene annotation, ortholog identification to tree inference, which can take many months to run—millions of CPU hours invested in this process are not uncommon—and specialised knowledge to oversee this process.
That's where Read2Tree comes in. Our new approach to tree inference bypasses the usual steps of genome assembly, annotation, and orthology inference. Instead, it uses existing knowledge of the protein sequence universe to directly reconstruct comprehensive sequence alignments from raw sequencing reads.
The approach is vastly faster than traditional methods and in many cases more accurate—the exception being when sequencing coverage is high and reference species very distant. Read2Tree is also flexible, working with genome and transcriptome, short and long reads, and sequencing coverage as low as 0.1x.
We were encouraged by the buzz the Read2Tree manuscript elicited on bioRxiv last year, and are delighted it has now been published in Nature Biotechnology.
What is Read2Tree good for?
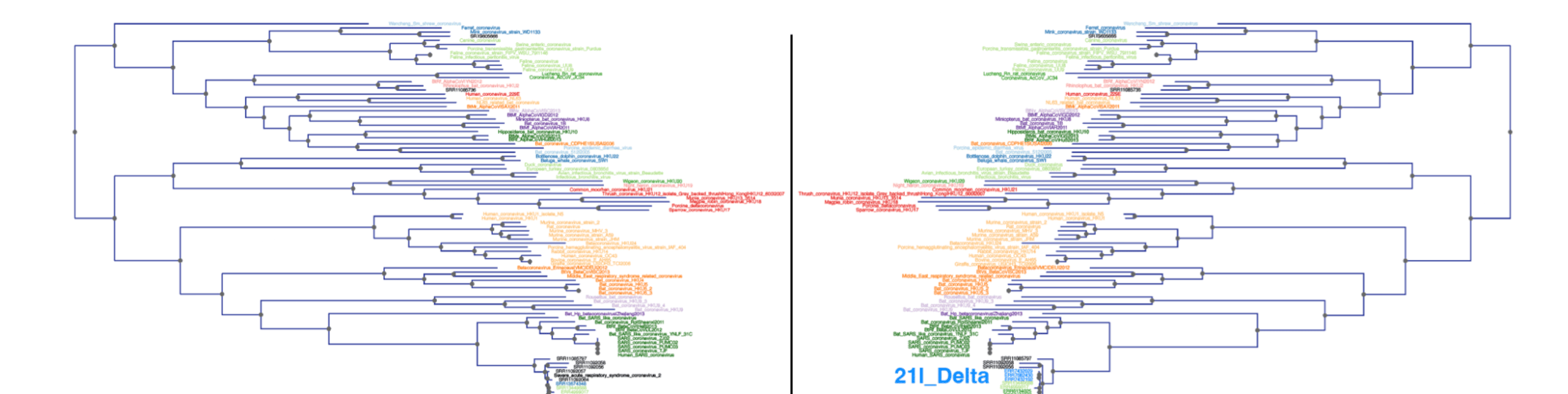
A nice illustration of Read2Tree’s potential was the reconstruction of a phylogeny of coronaviruses, which processed on the same tree diverse Coronaviridae sequences as well as 10,000 raw SARS-CoV-2 datasets from the Short Read Archive. The reconstructed tree was consistent with the lineage classification obtained from the UniProt reference proteomes, accurately recovering the main coronavirus genera and all subgenera (Figure 1). At the same time, the same phylogeny accurately clustered the sequences according to CDC variants of concerns classification. These results demonstrate the versatility and scalability of Read2Tree, making it suitable for both zoonotic surveillance and human epidemiology.

The ability to reconstruct phylogenetic trees from raw reads has additional advantages. Some genomes are deposited with poor or even entirely absent protein annotation sets. Processing genomes directly from raw reads can avoid this limitation and also decrease biases that arise from relying too heavily on specific reference genomes. Although some efforts have been made to "dehumanize" non-human great ape genomes, other clades still face similar biases that can be significantly reduced by processing raw reads.
Who might find it useful?
We think Read2Tree will be especially useful for small labs with limited bioinformatics expertise and computational resources, allowing them to perform state-of-the-art phylogenomics on particular species or environments of interest.
But it's not just small labs that can benefit from Read2Tree. Large consortia can also use it to regularly update their trees as new genomes are sequenced. This is especially important as more and more projects around comparative genomics are underway, such as the Earth BioGenome, the Darwin Tree of Life, or the European Reference Genome Atlas projects.
In addition, Read2Tree's ability to infer trees from much lower coverage than traditional methods means it can also be useful for quality control early in the process. This makes it a valuable tool for environmental and metagenomic applications, especially when combined with genome binning techniques.
Overall, Read2Tree is a powerful method for inferring phylogenetic trees directly from raw sequencing reads. We hope it will help make phylogenetic tree inference faster, more accurate, and more accessible to a wider range of researchers.
What’s next?
Now that the introductory Read2Tree paper is published, we are excited to explore new potential applications that we haven't been able to tackle so far. For instance, we have already received inquiries from researchers interested in using Read2Tree for ancient DNA applications or for monitoring systems that require fast turnaround time and low coverage.
Moving forward, we have two main goals. First, we aim to expand Read2Tree's capabilities to handle multi-species samples, which will enable an even broader range of applications in the metagenomics field. While long-read applications may offer the most benefit, we are confident that Read2Tree's ability to perform well with short-reads will also prove valuable in detangling multiple species.
Secondly, we plan to explore the use of Read2Tree in single-cell sequencing. This rapidly growing field involves sequencing individual cells, including cancer cells, and analysing their genetic information. Given Read2Tree's ability to operate with low coverage levels (down to 0.2x), we believe it could facilitate fast and accurate characterization of tumour or cell evolution.
We hope that Read2Tree will help streamline and democratise comparative genomics analyses. We are excited to see how researchers will apply this tool to further advance our understanding of genetics and evolution.
What’s the backstory?
Both of our labs (Fritz Sedlazeck’s and Christophe Dessimoz’s) have been collaborating for many years, and we've always enjoyed exchanging ideas even though our research interests are quite diverse. One of our interests over the years is how to combine our expertise in sequence analysis and ortholog comparison to develop new methodologies and gain new insights into biology.
It was during one of Fritz's visits to Christophe’s lab in Lausanne, Switzerland, that we started brainstorming ideas for a project that led to Read2Tree. Our goal was to overcome the limitations and bottlenecks of comparative genomics. We had some amazing cheese risotto, and the beautiful scenery fueled our discussions further (Figure 2).

David Dylus, the first author, was convinced that it was possible to bring our ideas to life, although he did not anticipate how much time and effort it would take (Figure 3). Even after he moved on to a new role in the pharmaceutical industry, he continued to work on Read2Tree after regular work hours. And when the COVID-19 pandemic hit, we had to face additional challenges, such as maintaining regular meetings and pushing the manuscript forward while not compromising on quality. We also faced technical issues, such as hard disk crashes and cluster updates that led to data loss, but David hang on.
Completing the paper was not an easy task, and one of the biggest challenges was organising and identifying all of the SRA data sets, including those related to yeast and COVID-19. Despite these challenges, we were able to bring the project to completion. It was a special joy to present the work at ISMB 2022, where Fritz and Christophe had the wonderful opportunity to meet in person, and we continued to discuss our work while enjoying good food and drinks by beautiful Mendota lake in Madison, Wisconsin.
In summary, nice food and lakeside views were instrumental in the making of Read2Tree.

Follow the Topic
-
Nature Biotechnology
A monthly journal covering the science and business of biotechnology, with new concepts in technology/methodology of relevance to the biological, biomedical, agricultural and environmental sciences.


![When PSMA-targeted therapy is not enough: high-risk localized prostate cancer after repeated [177Lu]Lu-PSMA radioligand therapy](/cdn-cgi/image/metadata=copyright,fit=scale-down,format=auto,quality=95,width=256,height=256/https://public-storage.zapnito.com/Ku6h7Yyp4Q0LXqRRMICCHR2v4LcOsmxMrmDPtOYuI1c)



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in