Tracking transmission of bacteriophages in the microbiome
Published in Microbiology

Foundational studies from Bäckhed[1], Feretti[2], Yassour[3] and others have shown that infants often acquire bacterial strains from the mother’s microbiome. These transmitted microbes can confer benefits, such as the ability to digest human milk oligosaccharides, a key source of nutrients in breast milk. Vertical transmission appears to be a key factor in establishing a healthy gut microbiome.
However, the gut microbiome is not just bacteria. Phages, or viruses that infect bacteria, are also present. Collectively called the “virome,” phages have been relatively understudied in the gut compared to bacteria, often going unnoticed in metagenomic sequencing experiments. More recently, we’ve started to understand the role phages play in the ecology of the microbiome, including functions such as driving bacterial evolution and horizontal gene transfer. (For a good review, see Shkoporov and Hill[4].)

CrAssphage is the preeminent example of a human gut bacteriophage. Although it’s present in the microbiome of most adults and can be abundant, it was discovered only recently, in 2014. Named for the cross-assembly process used to construct the first genome[5], crAssphage has been difficult to isolate and culture, with only one example of a culturable related-phage in the literature[6]. More recent analyses have shown that the prototypical crAssphage, or p-crAssphage, is just one member of a diverse group crAss-like phages[7]. To avoid ambiguity moving forward, I’ll use “p-crAssphage” to refer to the first characterized phage, and “crAss-like phages” to refer to the larger group
P-crAssphage and its relatives have captivated the Bhatt lab for years. The phages have evolved in our microbiome for millennia, and related phages have been discovered in the microbiome of non-human primates[8]. Even so, no associations with health or disease have been identified, and the specific bacterial host for p-crAssphage is still unknown (although accumulating evidence suggests host(s) in the genus Bacteroides)
In particular, our lab was really intrigued by a few big questions: Is p-crAssphage just a bystander in a healthy microbiome - a phage that replicates constantly without affecting the human host? Where do people typically acquire p-crAssphage? Can it be passed down from mother to child for generations? How many different phage strains does the average person have in their gut?
To begin to answer some of these questions, Fiona Tamburini, who has since graduated with her PhD, proposed looking in publicly available datasets that contained metagenomic sequencing of mother-infant pairs. We combed the literature, downloaded terabytes of data from the NCBI Short Read Archive, and eventually settled on two datasets: Bäckhed et al.[1] and Yassour et al.[3] Collectively, these datasets contained hundreds of mother and infant stool microbiome samples. Without the diligence and persistence of the authors who generated, analyzed, and made the data publicly available, our project would have never got off the ground, so I’m very grateful to them.
Searching for a metaphorical needle in a haystack
The first step was to identify which samples had crAss-like phages. To do this, we used the metagenomic classification program Kraken2[9], with a custom database that contained the assembled crAss-like phage genomes from Guerin et al.[7] This allowed us to see both the relative abundance of crAss-like phages, and the number of phage clusters that were present in a given sample.
We then had to define a threshold for presence of a crAss-like phage. This is a challenging problem with no consensus in the field, but these two options seemed most reasonable to us:
A relative abundance threshold, which selects samples with the highest crAss-like phage abundance, but may miss samples truly positive samples where bacteria make up the majority of the microbiome.
An absolute coverage threshold. A present phage should be detectable regardless of the abundance of other species, but an absolute threshold is obviously confounded by sequencing depth.
In the end, we chose an absolute cutoff of 1,000 reads mapping to the crAss-like phage cluster as evidence for presence, as this corresponds to roughly 1x sequencing coverage. This is also the threshold at which we started to assemble somewhat contiguous phage genomes.
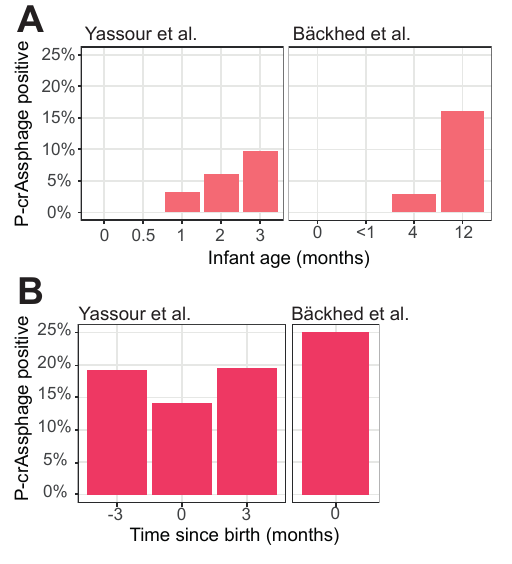
Using this threshold, we found that infants did not have crAss-like phages in their microbiome at birth; the earliest we detected them was in a sample from a three-day-old infant. The phages became increasingly common with age, but didn’t reach the levels observed in adult mothers by the end of sampling at one year. Interestingly, we found that adults could have up to eight of the ten crAss-like phage clusters detected in one sample.
Assembling identical genomes
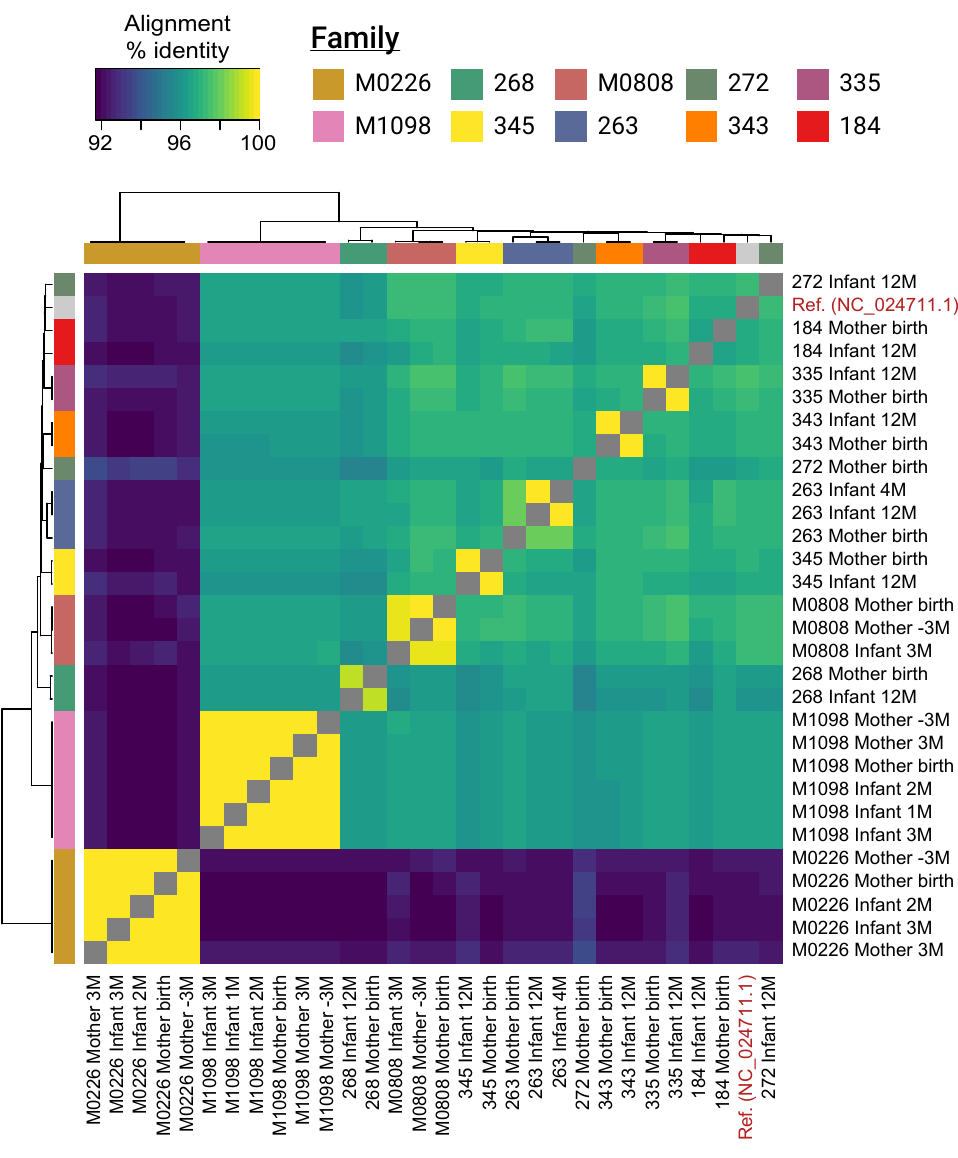
Next, we searched for evidence of mother-infant p-crAssphage transmission. We assembled the whole metagenome with metaSpades[10] and mapped contigs against the p-crAssphage reference genome. Then, we performed pairwise alignments of all well-assembled genomes. The result is what you see in Figure 1 of the paper, and reproduced here. Trust me when I say I was excited when this came out of the pipeline. In six out of ten cases, matched mother and infant samples had a nearly identical p-crAssphage genome, and no genomes from unrelated individuals were more than 96% identical. The clustering here was so strong, I was convinced transmission was playing a role.
Metagenomic assembly is a useful tool, but has drawbacks, including that it may “collapse” genomic regions that are not identical in closely related strains of the same species. We weren’t convinced that a simple assembly-based comparison was sufficient evidence for transmission, so we did a more thorough SNP-calling analysis to verify the results. When the strong clustering of mother-infant pairs held up to this more stringent analysis, I was convinced the research was headed in the right direction.
P-crAssphage strain diversity
After finding identical genomes in mothers and infants, we wanted to ask more in-depth questions about the p-crAssphage population in the microbiome. In particular, we wanted to know how many strains of p-crAspage an individual had, and if these numbers were different in mothers and infants.
To answer these questions, we mapped metagenomic sequencing reads to the p-crAssphage reference genome and looked for positions where multiple single nucleotide variants were maintained at intermediate frequency. Because these “multiallelic sites” aren’t fixed in the population, they give us information about the diversity of the crAssphage population in the microbiome.
Overall, we found that the p-crAssphage population in most individuals is less diverse than other phages in the microbiome, i.e., when you have crAssphage, you likely have a limited number of strains. This is in contrast to the bacterial portion of the microbiome, where multiple strains of the same species often coexist. Consistent with a bottleneck effect upon transmission, it appears that infants receive a further reduced diversity phage population from their mother.
We then looked at where multiallelic sites fell along the p-crAssphage genome, and what the predicted effects of the SNPs were. We found certain regions in the genome were “hotspots” for nonsynonymous (protein altering) changes, including genes encoding phage tail proteins. Tail proteins play a role in determining the bacterial hosts a phage can infect[11]. We hypothesized that maintaining a population with diverse tail proteins could be beneficial by expanding the host range of crAssphage, but further experiments involving isolate phages are necessary.
Barcode swapping can be a major confounder
The first version of the paper we posted to bioRxiv has a section that’s missing from the final publication. Using metagenomic data generated in our lab, we initially found identical p-crAssphage genomes among patients undergoing Hematopoietic Cell Transplantation (HCT) at Stanford hospital. This result was very exciting to us, as it suggested transmission in a new population. However, digging deeper into the results during the revision uncovered some inconsistencies that warranted further investigation. For example, some patients who shared p-crAssphage sequences never overlapped in their stays in the hospital, and some were even separated by multiple years.
We soon realized that pairs of samples with similar p-crAssphage genomes were always sequenced on the same lane of an Illumina machine. Comparing the multiplexing barcodes for these samples revealed that the suspicious sample pairs always shared one of the two multiplexing barcodes.
“Barcode swapping” was initially described by Illumina in 2017 and causes sequencing reads from one sample to appear like they belong to another sample sequenced on the same lane. Although barcode swapping typically only occurs at a rate of fractions of a percent, focusing on p-crAssphage actually made the problem worse for us. P-crAssphage reaches relative abundances as high as 20% in some samples. Even at the rate reported, thousands of p-crAssphage reads could swap from an abundant sample to a negative one. That would be poor coverage of a bacterial genome, but sufficient to assemble a p-crAssphage genome because the small genome size.
We’ve since moved to using unique dual indices in all multiplexed sequencing experiments. According to Illumina, this eliminates the issue of barcode swapping. We’re also much more aware of how the problem affects data previously generated in our lab.
The obvious next question is, “What about the other data you analyzed - couldn’t those findings be explained by barcode swapping as well?”
Unfortunately, information on barcode sequences was not reported in any of the publications we sourced data from, so we couldn’t quantify the magnitude of this effect. However, we believe our results, where we only observe genome sharing between family members, cannot be explained by barcode swapping alone. It is our view that multiplexing barcodes should be reported in all cases where publications generate sequencing data.
We removed the HCT patient section from the revised manuscript and admitted this confounding effect to the reviewers, who commended our choice to investigate the possibility of barcode swapping and appreciated that we were transparent with our findings. As we look forward to future experiments, it is important that we and others in the microbiome field who study transmission are aware of the impact of barcode swapping and other potential contributions to false positive findings, such as aerosolization of fluids and subsequent transfer of contents from one sample to another in multi-well plates.
Conclusions
Our data support the model that infants often acquire crAss-like phages from their mothers. However, there are some alternative hypotheses that we can’t rule out. Another housemate or other shared environmental source could be responsible for transmission. Collecting samples from other individuals is essential to understand the phenomenon completely.
Studying a phage in this project simplified many of the analyses. CrAss-like phage genomes assemble very easily out of metagenomic sequencing data, and the small genome size makes measuring strain diversity simple. While many methods have been developed to characterize strain diversity in bacteria, such as StrainPhlAn[12] PanPhlAn[13], our relatively simple read mapping approach worked without the need to identify marker genes.
Although we thoroughly characterized transmission and strain diversity of p-crAssphage, there are many areas for future research, including similar analyses of crAss-like phages. I hope other groups focus on these problems, especially using long-read sequencing[14] which would allow us to phase strain variants (and possibly capture entire crAss-like phage genomes in a single read).
I would like to thank my co authors for their work in preparation of the manuscript and this blog post, and the authors of the mother-infant papers for producing such valuable and rich datasets.
References
1. Bäckhed, F. et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 852 (2015).
2. Ferretti, P. et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 24, 133-145.e5 (2018).
3. Yassour, M. et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 24, 146-154.e4 (2018).
4. Shkoporov, A. N. & Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 25, 195–209 (2019).
5. Dutilh, B. E. et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun 5, 4498 (2014).
6. Shkoporov, A. N. et al. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 9, 4781 (2018).
7. Guerin, E. et al. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 0, (2018).
8. Edwards, R. A. et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 1 (2019) doi:10.1038/s41564-019-0494-6.
9. Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 1–13 (2019).
10. Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
11. Sordi, L. D., Khanna, V. & Debarbieux, L. The Gut Microbiota Facilitates Drifts in the Genetic Diversity and Infectivity of Bacterial Viruses. Cell Host Microbe 22, 801-808.e3 (2017).
12. Truong, D. T., Tett, A., Pasolli, E., Huttenhower, C. & Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 27, 626–638 (2017).
13. Scholz, M. et al. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods 13, 435–438 (2016).
14. Moss, E. L. & Bhatt, A. S. Generating closed bacterial genomes from long-read nanopore sequencing of microbiomes. bioRxiv 489641 (2018) doi:10.1101/489641.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in