Triggering nanofiber nucleation upon heating

Microfilaments and microtubules in cells fulfill a variety of functions including controlling cell movement, changes in cell shape, and molecular cargo transport. To carry out these dynamic functions the filaments and tubules must be transformable: they need to elongate and shorten, assemble and disassemble. What makes this possible is the requirement of energy input to drive their assembly. Similarly, some natural fibers such as collagen have been shown to require heat input to trigger their self assembly when studied outside the cell1.
Most synthetic supramolecular fibrous systems rely on directional hydrogen bonding to guide their formation; however, these intermolecular interactions are static and decrease in stability as temperature increases. Building synthetic systems that resemble biological ones requires a different set of design parameters.
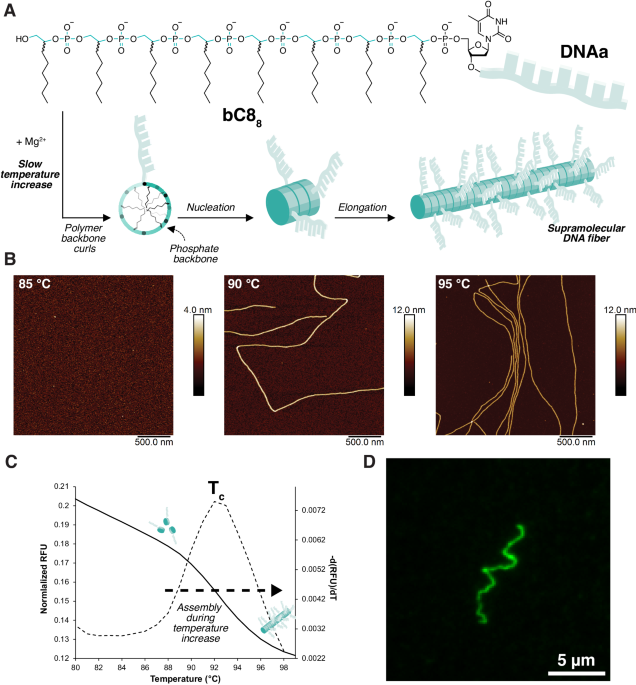
At the end of my PhD, my departure was delayed due to the lingering pandemic. Thankfully, my supervisor Prof. Hanadi Sleiman was incredibly generous and supported my stay as long as necessary. This additional time allowed me the luxury to investigate some more unusual findings from my thesis that we had previously been unable to completely interpret. One of these was the discovery of a series of DNA-polymers that broke the rules of traditional block copolymer theory2. Increasing their hydrophobic character caused their self-assembled morphology to transition from long nanofibers (Figure 1) to very short cylinders and spheres – the opposite trend as predicted. We also found that the heating rate was critical for controlling the length of the fibers, rather than the cooling rate, and that heating drove fiber formation through a nucleation-growth mechanism.
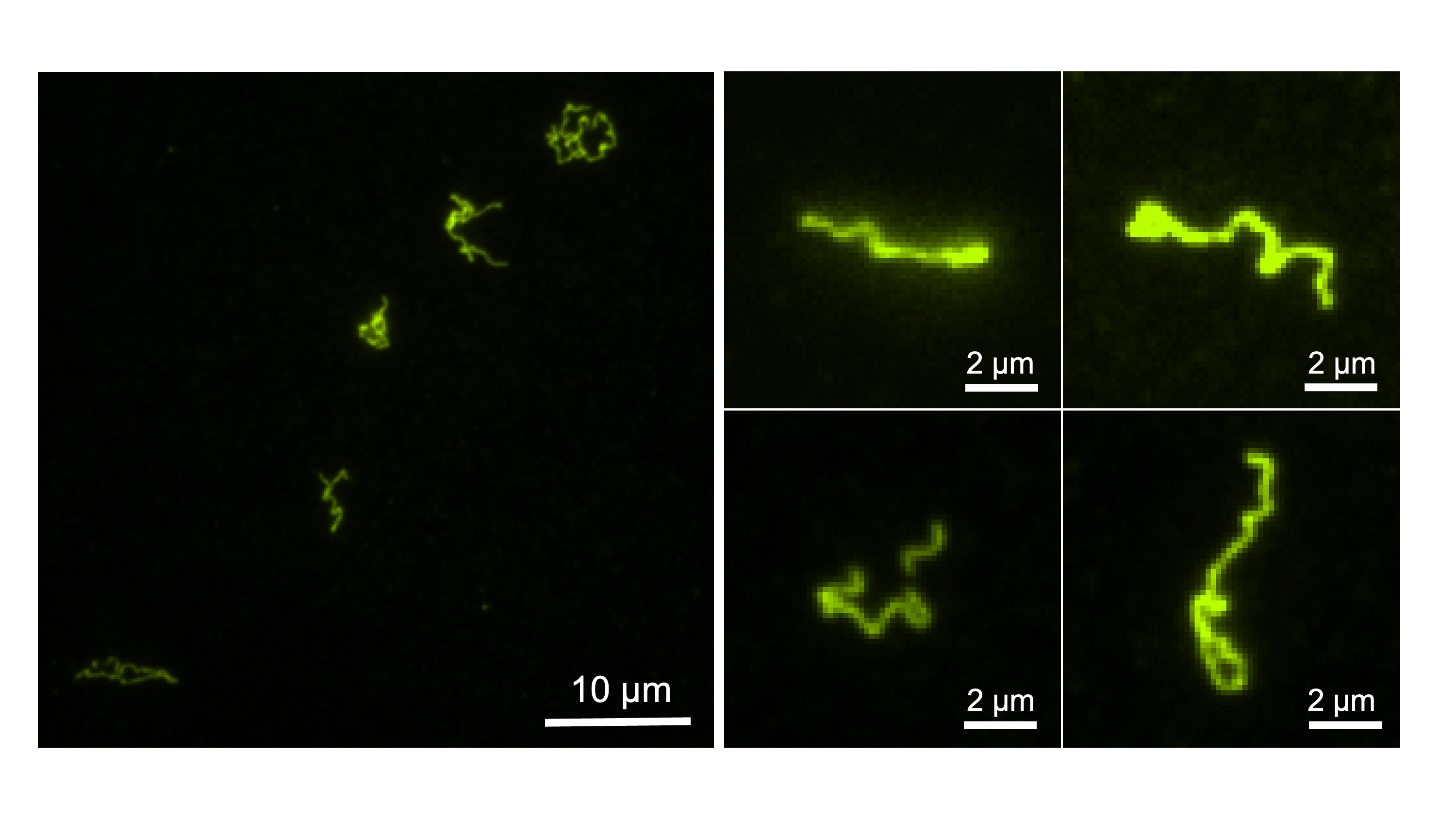
Through the contributions of various colleagues and collaborators, we found that the explanation for this unusual behaviour was the requirement for the DNA-poymer to assume a specific conformation in order to form fibers. Heating provided the necessary energy input to convert the DNA-polymer from a globular state to a structured one. At room temperature the DNA-polymer was compeltely soluble in aqueous solution, but upon heating nanofibers remarkably nucleated and elongated, growing longer as the temperature increased beyond 90 °C (Figure 2). Further, we found that the propensity to form the high-energy conformation was dependent on the DNA-polymer length: more hydrophobic character was geometrically unsuited for fiber-formation.

So if heat was required to drive the nanofiber formation, what happened when the fibers returned to room temperature? We found that the nanofibers were metastable: once cooled they slowly disassembled and mixed. In this way, our system resembled biological ones, where the default state is unassembled, and energy is needed to grow fibers when desired. Another parallel we saw between our DNA polymer and biological systems was the need for the DNA-polymer to assume a specific conformation, much like polymer folding.
Finally, we showed that the metastable fibers could be stabilized through the addition of a small molecule that formed a network of hydrogen bonding between the DNA sequences on the outside of the fibers. In this way we showed that the DNA segment of the fibers has utility by virtue of its highly specific molecular structure.
We predict that our system and ones like it will find applications in materials such as stimuli-responsive biomaterials and soft robotics as a supramolecular analogue of lower critical solution temperature (LCST) polymers.
References
- De Greef, T. et al. Supramolecular polymerization. Chem. Rev. 109, 5687–5754 (2009).
- Israelachvili, J. N. Intermolecular and Surface Forces, 3rd Edition. (Elsevier Science, 2011).
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in