Using genome structure, rather than gene trees, to infer relationships in ancient allopolyploid lineages
Published in Ecology & Evolution

That hybridization should be followed by genome duplication is plausible because a hybrid that undergoes a duplication of its two sets of chromosomes can immediately restore fertility, since each chromosome at meiosis will have a single homologous chromosome to pair with. If the parents of an allotetraploid hybrid were relatively different from each other, the two parental genomes may effectively coexist and maintain heterozygosity over generations. Such allotetraploid lineages may thrive and undergo speciation, with descendants eventually experiencing further hybridization or introgression. As long recognized, the coexistence in each allotetraploid individual of both homologous chromosomes, which have similar gene composition, and homeologous chromosomes, which have arisen from the genome doubling, poses problems for the reconstruction of species relationships. This is because species relationships are ideally inferred only from orthologous loci, that is, genes related through speciation events. Identifying such orthologs in polyploids, however, is difficult because gene duplication obscures the relationships among genes. When duplicated genes are mistaken for orthologs, gene tree topologies often differ from the species tree topology (Smith and Hahn 2022, for a review).
The doctoral research project of Ya-Mei Ding on the phylogeny of the walnut family, Juglandacaeae, faced this problem because the family is an ancient allopolyploid lineage. The walnut family comprises just 63 species in eight genera (Fig. 1), but includes many of the World’s commercially most valuable nut-producing crops, such as Persian walnut, Chinese Iron walnut (genus Juglans), pecan, and hickory (genus Carya). Ten phylogenetic studies over the past 20 years have obtained contradicting topologies for deep nodes in this family. Ya-Mei and her advisors, Da-Yong Zhang and Wei-Ning Bai at the College of Life Sciences, Beijing Normal University in Beijing, decided to try a new approach that would use genome structure to infer organismal relationships in the Juglandaceae. For this, they took advantage of five available chromosome-level genome assemblies and newly assembled the genomes of two key representatives, Engelhardia roxburghiana and Rhoiptelea chiliantha, the latter sister to all other Juglandaceae (Fig. 1).
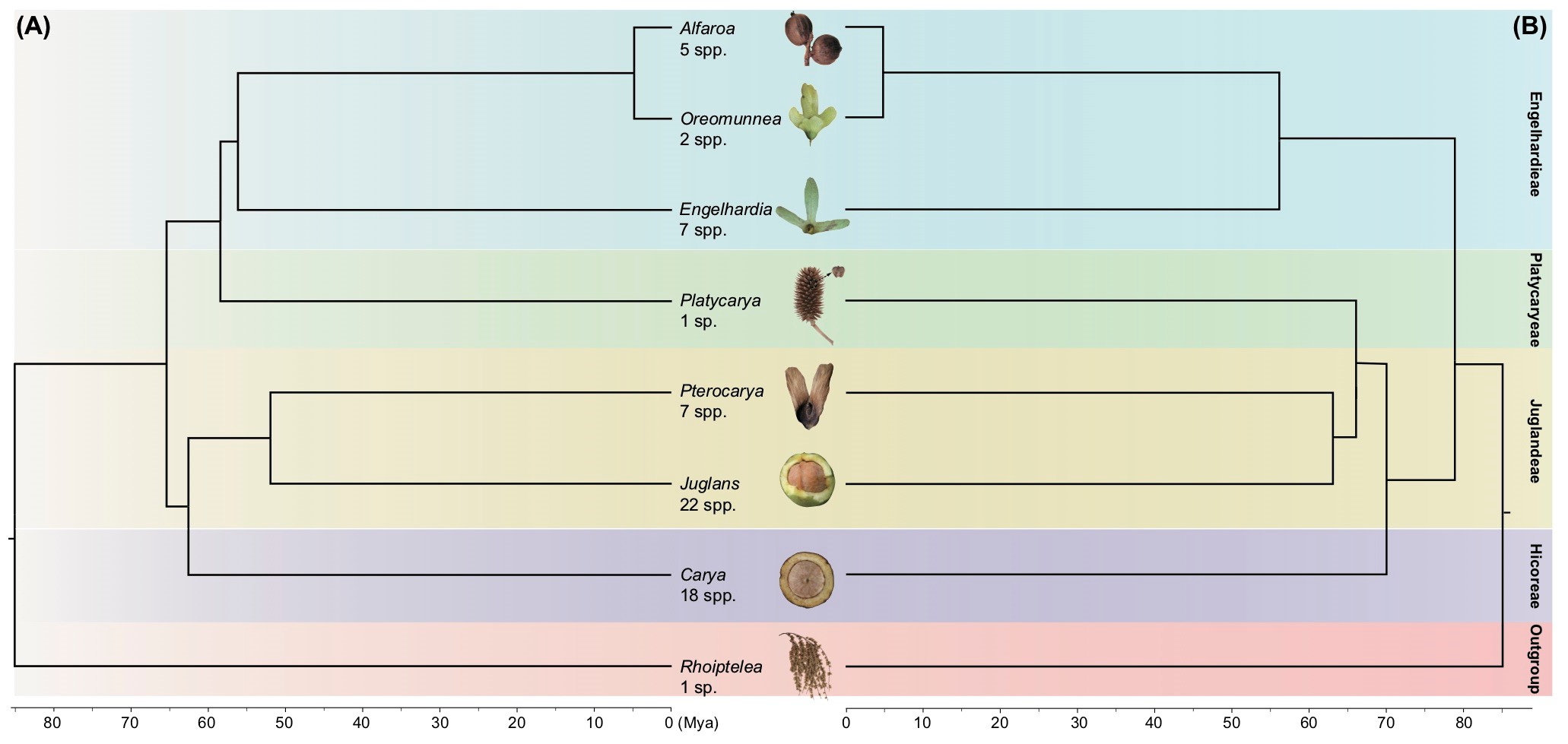
Fig. 1 (A) The morphology-based topology for the Juglandaceae obtained by Wing and Hickey (1984) and (B) one of many DNA-alignment based phylogenies. Despite numerous morphological and molecular studies, the placement of the single species of Platycarya has remained unclear since the mid-1980s.
Based on the number of retained ancestrally inherited gene on chromosomes, i.e., genes also found in Quercus lobata, a representative of Fagales to which Juglandaceae belong, Ya-Mei could assign each of the homoeologous chromosomes of the seven Juglandaceae to one of 14 subgenomes. For lack of a better term, the two coexisting subgenomes were called ‘dominant’ and ‘recessive.’ The corresponding pairs of homoeologous chromosomes were additionally verified by intraspecific collinear blocks. The presence or absence of genes and the microsynteny along chromosomal blocks were then used as phylogenetic data. Figure 2 illustrates the workflow. It began by detecting synteny clusters using a well-known network clustering algorithm (Infomap), and clusters were then transformed into a binary presence-absence matrix where rows and columns represent species and clusters, respectively. This binary matrix was then used to infer a maximum likelihood phylogeny with IQ-TREE 2, using the MK+FO+R model appropriate for morphological data matrices, with optimized state frequencies and freely distributed rates (Minh et al. 2020; Fig. 2-6).
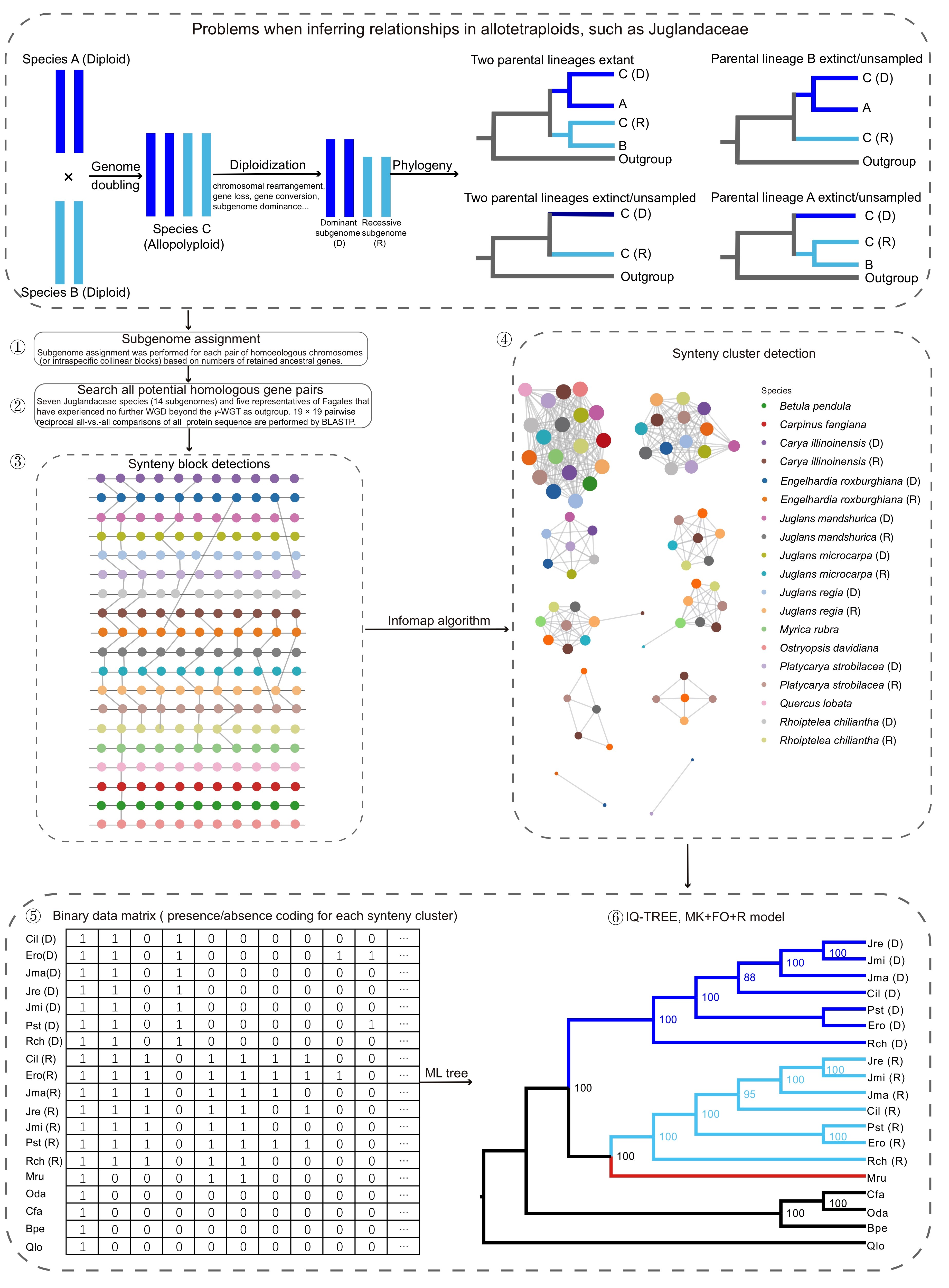
Fig. 2. Illustrating the pipeline for extracting phylogenetic signal from genomic structure.
Surprisingly, the genus phylogeny obtained from both the chromosomal microsynteny data and the local gene content data differed in one deep node from the one obtained from DNA-sequence alignments. Different parameter settings, involving the minimum number of collinear gene pairs and the maximum number of intervening genes between adjacent blocks, did not change the topology. The conflicting deep node concerned the enigmatic east Asian genus Platycarya with the single species, P. strobilacea. Based on its genome structure, this species is placed as found in a 1984 morphology-based study (Wing and Hickey, 1984), but based on gene trees it is instead placed as in alignment-based topologies (as illustrated in Fig. 1).
While further genome assemblies from other Juglandaceae, especially two small Mexican genera, Alfaroa and Oreomunnea (Fig. 1), may help to further clarify the family’s genus phylogeny, simply on theoretical grounds, phylogenies for allotetraploid lineages are probably best inferred from genome-structural data, rather than gene coalescence trees, which rely mostly on signal from DNA substitutions. This is partly a matter of genome-structure data permitting to distinguish coexisting homeologous chromosomes and to use them separately to infer organismal ancestry. More importantly, any large genome-structural variants capable of altering gene order and/or content probably rarely travel horizontally between species, thus making tree inference based on gene order or content more robust to introgression.
References
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., et al. 2020. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37:1530–1534.
Pett, W., Adamski, M., Adamska, M., Francis, W. R., Eitel, M., et al. 2019. The role of homology and orthology in the phylogenomic analysis of metazoan gene content. Mol. Biol. Evol. 36:643-649.
Smith, M. L., and Hahn, M. W. 2022. The frequency and topology of pseudoorthologs. Syst. Biol. 71:649–659.
Wing, S. L., and Hickey, L. J. 1984. The Platycarya perplex and the evolution of the Juglandaceae. Am. J. Bot. 71:388–411.
Zhao, T., Zwaenepoel, A., Xue, J. Y., Kao, S. M., Li, Z., et al. 2021. Whole-genome microsynteny-based phylogeny of angiosperms. Nat. Commun. 12:3498.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in