Deep into the mysterious world of giant viruses: finding the cryptic mirusviruses in metagenomic “taxonomic blind spots” of Tara Oceans and beyond
Published in Ecology & Evolution, Microbiology, and Genetics & Genomics
If there was a contest for “environmental genomes most challenging to extract from metagenomes”, the cryptic mirusvirus genomes would certainly be well positioned, and this despite the remarkable legacy of genome-resolved metagenomic surveys completed in the last two decades. This behind the paper blogpost tells the story of how segments of metagenomic assemblies (metagenomic bins) containing an evolutionary constrained hallmark virus gene but otherwise almost entirely lacking protein-level sequence similarity to anything known to date (taxonomic blind spots) provided crucial guidance to identify vast evolutionary landscapes of large and giant eukaryotic DNA virus genomes within the recently identified phylum Mirusviricota. In other words, it is a near-complete lack of taxonomic signal that was ultimately used as the main signal to delineate and extract cryptic mirusvirus genomes at a global scale.
Mirusviruses and the nucleus of planktonic unicellular eukaryotes
Mirusviruses represent a diversified phylum (Miruviricota) of large and giant DNA viruses prevalent in aquatic ecosystems where they infect unicellular eukaryotes. Their discovery not only filled an important evolutionary gap between phages and herpesviruses but also demonstrated that not just one (the phylum Nucleocytoviricota and its highly publicized giant viruses such as mimivirus and pandoravirus) but two major phyla of eukaryotic DNA viruses prevail in the surface of the oceans and seas. In our recent Nature Microbiology publication (with Ulysse Guyet and Sofia Medvedeva as the two talented co-first authors), we expanded the genomic landscape of Miruviricota and identified three major putative orders, one minor putative order as well as 13 cryptic putative orders. We provided multiple lines of evidence strongly indicating that many mirusviruses replicate in the nucleus of planktonic unicellular eukaryotes, contrasting with nucleocytoviruses that for the most part are predicted to replicate in the cytoplasm. With the publication focusing almost entirely on the major putative orders, this blogpost tells the story of how metagenomic “taxonomic bling spots” guided us towards the genomic landscape of most of the cryptic putative orders.
Metagenomic “taxonomic blind spots” and the cryptic mirusviruses
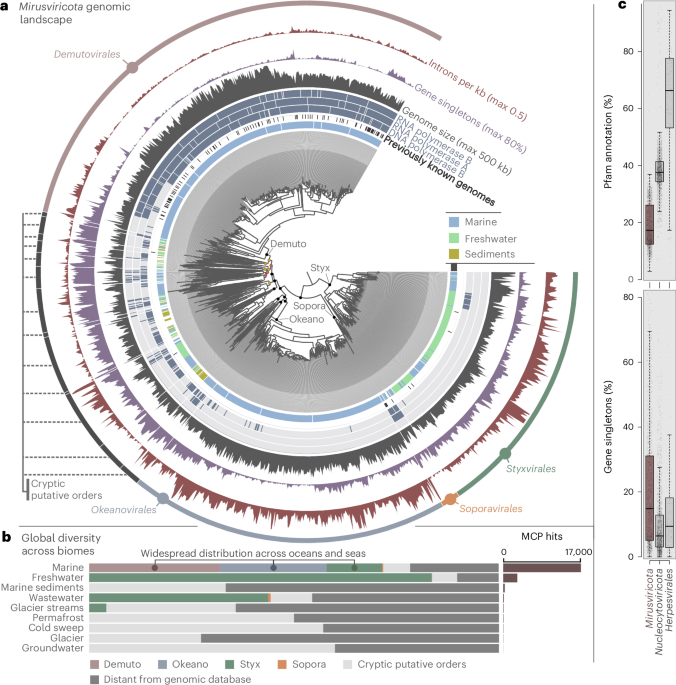
Genome-resolved metagenomics aims at finding genomic fragments (“contigs”) belonging to the same environmental genome within a metagenomic assembly. A classic genome-resolved metagenomic workflow has two main steps. First, relevant biological information (sequence composition and differential coverage) delineates environmental genomes from the metagenomic assembly (“resolving the metagenomic puzzle” step). Second, completion and redundancy scores are computed for each environmental genome using a relevant single copy core gene collection, with the scores being used as a proxy of genomic quality (“how well was the metagenomic puzzled resolved” step). Starting in 2019, we performed four distinct genome-resolved metagenomic surveys (thereafter named screening phases) dedicated to Mirusviricota, each of them providing critical environmental genomic contributions, and with only the last one allowing the characterization of most cryptic mirusviruses (Figure 1). Among the researchers actively involved in these screening phases, Morgan Gaïa, Sofia Medvedeva, Hans-Joachim Ruscheweyh, Shinichi Sunagawa, Eric Pelletier, Patrick Forterre and Mart Krupovic must be mentioned for their critical roles. A special point to Morgan Gaïa for pointing at the evolutionary prominence of viral polymerases (the starting point of this journey) and to Mart Krupovic for finding the gene encoding the major capsid protein (MCP) of mirusviruses, which is critical to the making of the viral particle and highly unexpectedly, connected them to herpesviruses and not nucleocytoviruses evolution-wise. But when it comes to the cryptic mirusvirus genomes, it is Ulysse Guyet that unlocked their characterization by allowing the effective identification of taxonomic blind spots across metagenomes.

Figure 1: Four screening phases to recover the genomic landscape of Mirusviricota. Maximum-likelihood phylogenomic tree of 1,204 Mirusviricota genomes based on the concatenation of manually curated alignments of MCP, terminase and portal proteins (1,871 amino acid positions). Details about the tree can be found in the Figure 1 of our Nature Microbiology publication. The four first layers indicate the screening phase origin of genomes present in the final non-redundant genomic database of Mirusviricota. Next layer indicates genomes that are single representatives of a putative family. Finally, the last layer displays the quality score (completion minus redundancy) of genomes for which a single copy core gene collection is available. Selections indicate the major putative orders as well as the cryptic putative orders.
Screening phase 1: A first major putative order of mirusviruses.
In 2019, large Tara Oceans metagenomic co-assemblies processed by the bioinformatic platform anvi’o had already been used to characterize environmental genomes abundant in the sunlit oceans and corresponding to Bacteria, Archaea and Eukarya. At that point, we started searching for giant eukaryotic viruses of the phylum Nucleocytoviricota. In this first screening phase, we used a phylogeny-guided genome-resolved metagenomic approach focused on the RNA polymerase gene (a hallmark gene of most nucleocytoviruses) and found deep-branching clades that led to the manual genomic characterization of the mirusvirus major putative order Demutovirales. The discovery of demutoviruses, a marriage of data-driven science and pure luck, triggered a colorful adventure exploring the phylum Mirusviricota.
Screening phase 2: Two additional major putative orders of mirusviruses.
Core genes among the Demutovirales genomes, in the context of 3D structure predictions, led to the identification of the mirusvirus MCP. In our second screening phase, we used this hallmark gene as guidance to expand the scope of our explorations within the same Tara Oceans metagenomic co-assemblies. Still within anvi’o, we manually characterized additional genomes that for the most part correspond to two previously overlooked major putative orders: Okeanovirales and Styxvirales. This phase provided the first indication that most mirusviruses lack any polymerase. If it was not for Demutovirales, we would have entirely missed the mirusviruses in our first screening phase. After completing this screening phase, sufficient genomic material was available to identify some of the building blocks in the genomic fabric of each major putative order. With that information, we created medium-efficacy single copy core gene collections and roughly assessed genomic quality scores. These collections are now obsolete but were critical at the time to gain confidence and move in the right direction.
Screening phase 3: navigating metagenomes with taxonomic signal as guidance.
Anvi’o and Tara Oceans offered an effective, albeit restrictive environment to delineate, visually explore and gain confidence in the biological relevance and quality of mirusvirus genomes. In our third screening phase, we moved away from this safe environment to embrace a global database (the mOTUs resource, by collaborating with Hans-Joachim Ruscheweyh and Shinichi Sunagawa) providing more than 100,000 metagenomic assemblies across a wide range of ecosystems along with automatically generated bins. We identified and subsequently focused on more than 2,000 metagenomic bins containing a least one mirusvirus MCP. Most of the MCP-positive metagenomic bins originate from aquatic ecosystems. For automatic decontamination purposes, we created a curated protein database covering major cellular and viral lineages (Bacteria, Archaea, Eukarya, chloroplasts, Mirusviricota, Nucleocytoviricota) and used it to assign a high-ranking taxonomy to contigs, as well as family-level taxonomic assignments within Mirusviricota. For each MCP-positive metagenomic bin, all contigs affiliated to the same mirusvirus family were placed into a mirusvirus bin. During this process, highly relevant single copy core gene collections (often down to a single family) were built to assess the quality of each mirusvirus bin, and those representing high-quality environmental genomes were harvested. With this automatic binning and decontamination approach, we dramatically expanded the number of high-quality mirusvirus genomes (Figure 1). At that point, we were able to characterize and assess the quality of most mirusvirus genomes as effectively as one would commonly do for cellular genomes. No more walking in the dark when it comes to mirusviruses, we thought. Our fourth and last screening phase proved us wrong.
Screening phase 4: MCP-containing metagenomic taxonomic blind spots.
Looking back at the MCP-positive metagenomic bins, we noticed that many of them had a size coherent with a mirusvirus genome (200-500 kb) and yet did not contain any contig-level taxonomic signal, whether it was for Bacteria, Archaea, Eukarya, chloroplast, Nucleocytoviricota or Mirusviricota. With a complete lack of taxonomic signal among bins containing a critical hallmark gene for mirusvirus, we speculated that they must represent genomes corresponding to one or multiple deep-branching mirusvirus lineages that almost entirely lack protein-level sequence similarity to genomes characterized during the first three screening phases. Realizing that the lack of taxonomic signal could be used as the main signal to delineate environmental genomes, we extracted such contigs and labelled them as cryptic mirusvirus genomes. While our subsequent investigations confirmed the biological relevance of these genomes, we still lack enough genomic material to build effective single copy core gene collections for most of them (Figure 1). Thus, we still navigate in dark waters when it comes to the cryptic mirusvirus genomes, and numerous genomes are needed to provide the desired single copy core gene collections. It is unclear how many collections will be required for the entirety of Mirusviricota, but current trends suggest it might be well over one hundred. For context, a single collection is needed for all of Bacteria.
A brief perspective on the cryptic mirusviruses
After completion of the four screening phases, a database of 1,257 non-redundant mirusvirus genomes was built and used to produce a taxonomic framework for Mirusviricota. With this framework, the major putative orders (nearly 1,000 genomes) are organized into 30 distinct putative families, 6 of which being represented by a single genome. In contrast, the 13 cryptic putative orders (just over 300 genomes) are organized into 134 putative families, 96 of which being represented by a single genome (Figure 1). It is striking that nearly one third of the cryptic mirusvirus genomes characterized thus far correspond to single representatives of entire viral families. If the cryptic putative orders were to represent a vast and dense forest, then what we have done so far is the identification of very few distantly related trees. The randomly sampled trees tell us that there must be a vast and dense forest, but we just do not see it yet. Despite this gross under sampling situation (driven in part by their rarity in metagenomic datasets), the cryptic mirusvirus genomes already encapsulate most of the captured evolutionary diversity of Mirusviricota. Thus, we anticipate that at least a few additional mirusvirus putative orders, and certainly hundreds of additional families will be discovered in the coming years (at a pace following the growth of publicly available metagenomes), expanding the scope of the known diversity of large and giant eukaryotic DNA viruses.
This blog is dedicated to the many people involved in the Tara Oceans expeditions, development of the anvi'o platform, production of publicly available metagenomes in general, and finally the building of the mOTUs resource. You all contributed to this journey deep into the mysterious world of mirusviruses. Thank you.
Follow the Topic
-
Nature Microbiology

An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.
Related Collections
With Collections, you can get published faster and increase your visibility.
The Clinical Microbiome
Publishing Model: Hybrid
Deadline: Mar 11, 2026
Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in