How viruses trick the immune system to escape antiviral defences
Published in Microbiology

The eradication of smallpox is one of the greatest achievements of humankind. This feat was achieved through a global vaccination campaign led by the WHO using vaccinia virus, a virus of the same genus as variola virus, the cause of smallpox. After smallpox eradication in 1980, virologists and immunologists sought to understand what made vaccinia such a good vaccine, and how this knowledge might be exploited to improve other vaccines and further develop vaccinia as a vaccine vector against other diseases. Our lab has undertaken this task for decades by investigating how vaccinia virus interacts with our immune system.
Like predators and preys, viruses and their hosts coexist in a struggle for survival. Hosts deploy immune defences to fight and eliminate invading viruses, and in response viruses evolve countermeasures to evade or overcome these defences, thereby allowing them to replicate and spread to new hosts. When our cells sense a virus infection, they express proteins that are antiviral or promote inflammation, a process by which the host responds to the infection and activates the immune system. NF-κB is an important transcription factor that is activated in response to both virus sensing and inflammatory molecules.
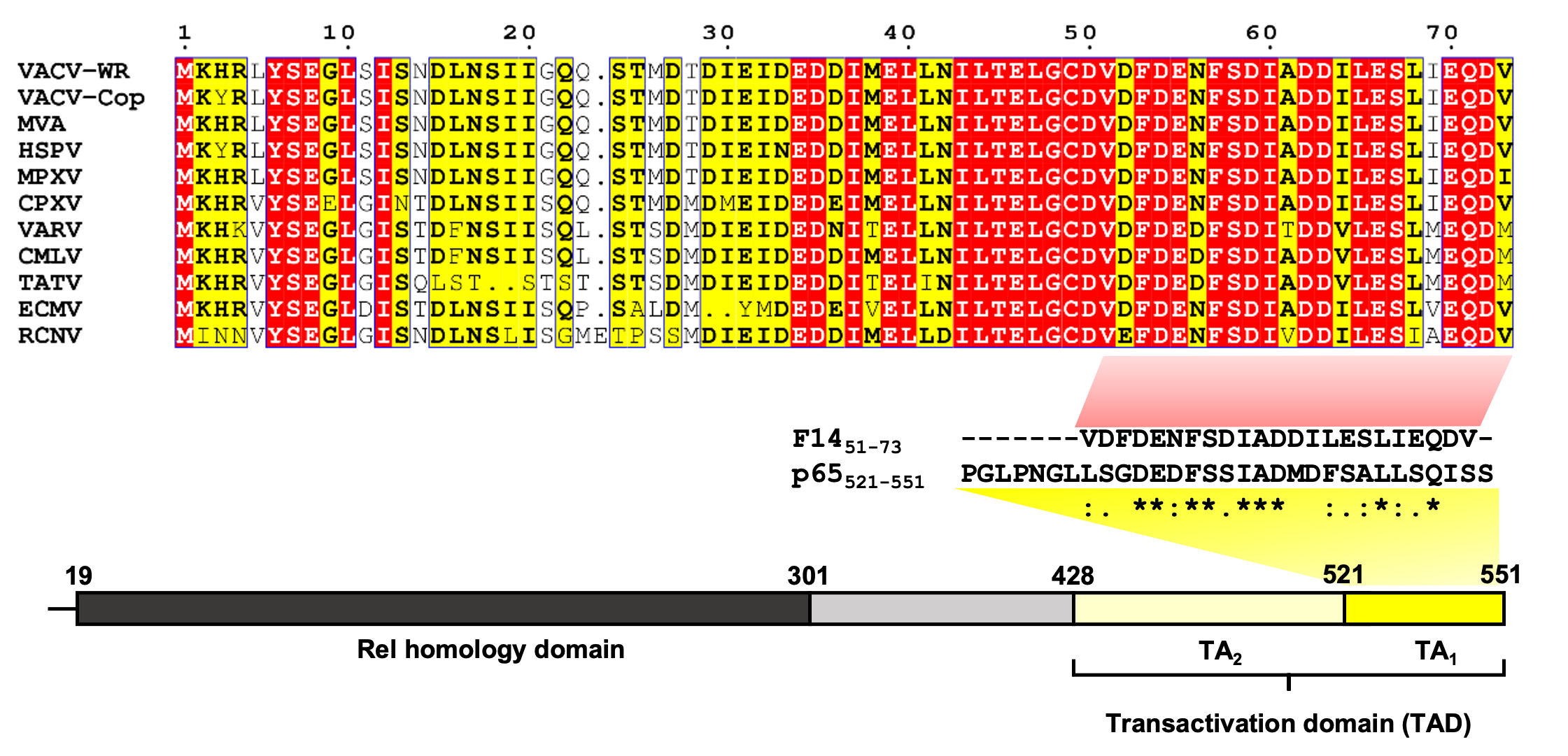
The importance of NF-κB is illustrated by the fact that vaccinia virus makes >10 different proteins that each antagonise NF-κB. In 2014, work from our lab predicted that vaccinia virus encodes at least one additional NF-κB inhibitor and when Jonas joined our lab he sought and identified this inhibitor as protein F14, a small protein of only 73 amino acids. Despite the presence of >10 other NF-κB inhibitors, F14 is important for the virus in vivo and contributes to virulence. Consistent with this, F14 is conserved among other viruses in the same genus as vaccinia virus (the orthopoxviruses). These include cowpox virus, a zoonotic virus that, despite its name, causes sporadic outbreaks in zoo and pet animals and humans across Europe; monkeypox virus, another zoonotic virus that circulates in Central and West Africa and hit the news when rare cases are imported into North America or Europe; and variola virus. Even ancient variola virus sequences retrieved from human teeth from the Viking era (10th century) encode highly conserved orthologues of F14. This suggests that F14 exerts an important function during virus infection.
In hindsight, defining the physiological relevance of F14 was straightforward, but what about its mechanism of action as an inhibitor of NF-κB? From the beginning, we knew that F14 acted in the nucleus, after NF-κB translocated from the cytoplasm to the nucleus to activate gene expression. However, a watershed moment in understanding how F14 functioned was analysing the F14 amino acid sequence on Phyre2, an algorithm that predicts the 3-dimensional fold of proteins (AlphaFold did not exist back in the days). Phyre2 predicted that the C-terminal third of F14 adopts a fold very similar to the region of the cellular protein p65 that interacts with another protein called CBP. p65 is the subunit of NF-κB responsible for activating the transcription of the target genes when NF-κB binds the DNA. The region of p65 that F14 resembles is the transactivation domain (TAD), meaning the region of p65 that switches on the transcription of NF-κB target genes, and this region of p65 shared significant amino acid sequence with F14.
The similarity of F14 with p65 TAD caused us to hypothesise that F14 might interact with the same proteins that interact with p65 TAD, like CBP. Indeed, F14 interacts with CBP, and disrupts the interaction between CBP and p65. When we mutated the C terminus of F14 that shares similarity with p65 TAD, the mutants with reduced interaction with CBP also lost the ability to inhibit NF-κB. This led us to conclude that F14 was mimicking the p65 TAD.
Molecular mimicry is a widespread phenomenon in host-pathogen interactions. Mimicry also takes place in higher levels of organisation of the biosphere. Owl butterflies, walking stick insects, bee orchids and orchid mantises are just some popular examples. Mimicry was noticed by Charles Darwin and in 1871 he wrote to Dr Fritz Müller saying, “No part of your letter has interested me more than that about mimicry”. Müller was a German migrant and naturalist living in Southern Brazil who had extensive correspondence with Darwin and became an early advocate of Darwin’s theory of evolution by natural selection. In Brazil, Müller observed many species of unpalatable butterflies that mimic each other to escape predation. This biological phenomenon caught Darwin’s attention because Müller believed that mimicry had arisen through natural selection. If mimicry was interesting for Darwin, it was interesting for us, so we investigated the nitty gritty of how F14 worked.
We found that by disrupting p65-CBP interaction, F14 was interfering with the molecular events necessary for the transcription of genes regulated by NF-κB that promote inflammation. CBP is an acetyltransferase, an enzyme that adds an acetyl group to lysine residues of other proteins. p65 is one of them and, once acetylated, p65 recruits the protein BRD4 to target gene promoters, where BRD4 promotes gene transcription (see Figure 1).
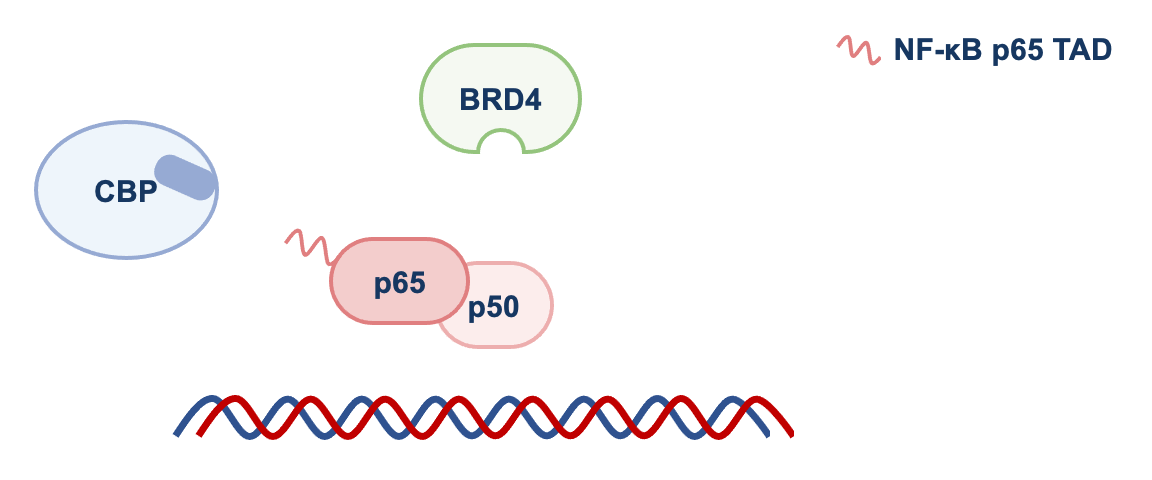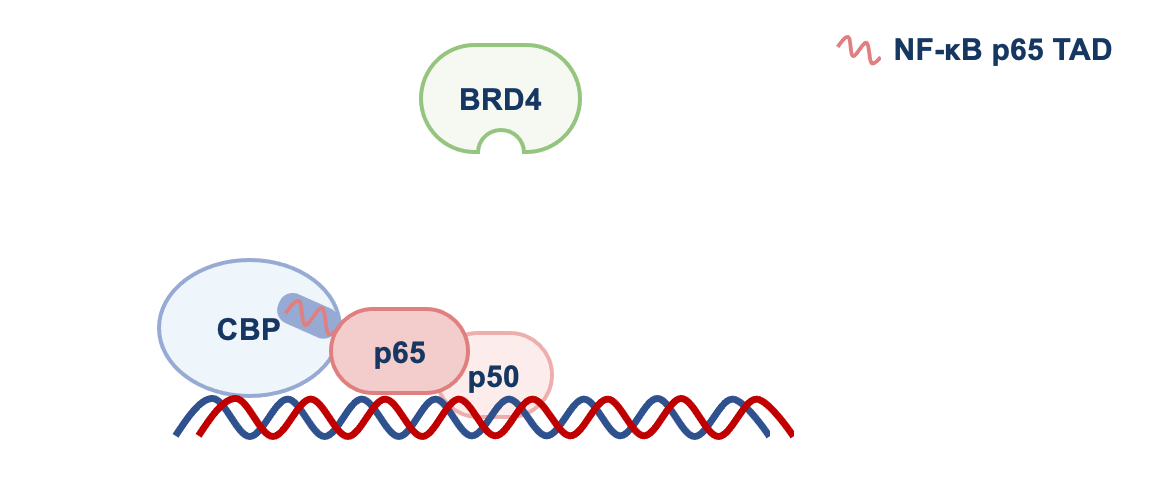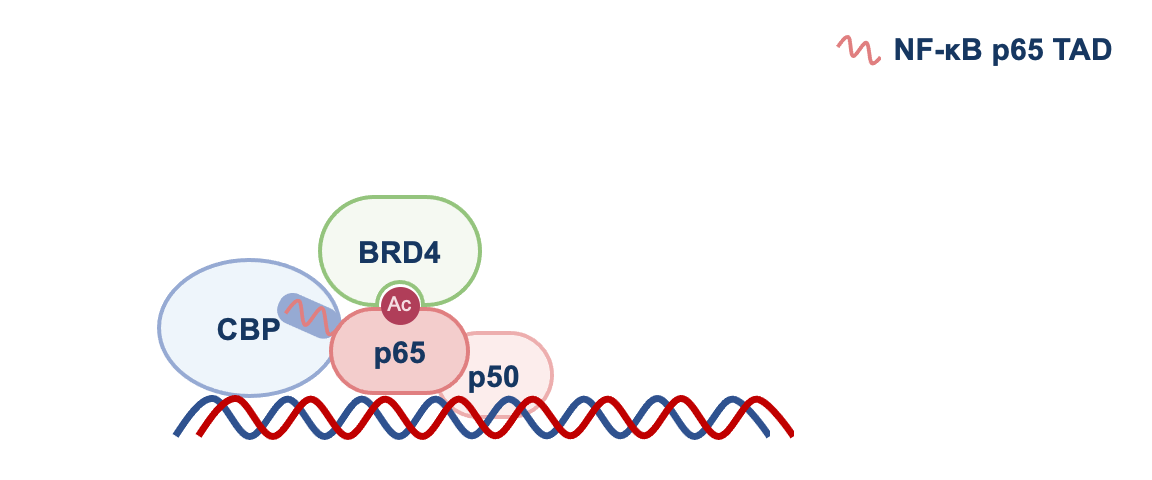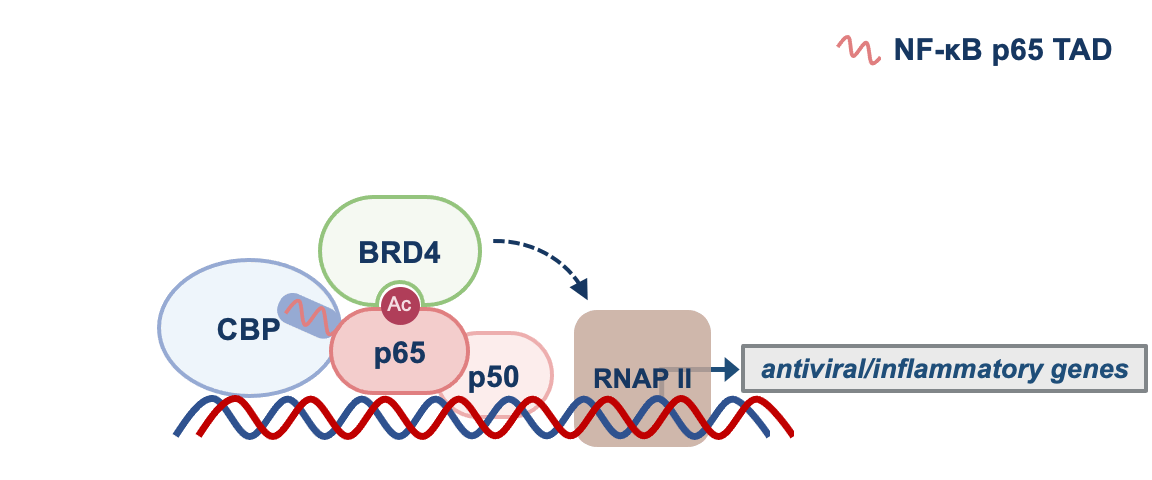
NF-κB = p65 and p50. TAD = transactivation domain. RNAP II = RNA polymerase II. See text for more details.
So, to summarise so far, F14 mimics the p65 TAD, disrupts p65 interaction with CBP, inhibits the acetylation of p65 and therefore the recruitment of BRD4 to gene promoters regulated by NF-κB. But here it got even more interesting because we found F14 was an inhibitor of some but not all NF-κB-regulated genes. Unlike other viral inhibitors of NF-κB, F14 is a selective inhibitor. To understand how F14 was selective, we studied the recruitment of BRD4 to NF-κB-regulated promoters and found that F14 inhibited BRD4 recruitment to some, but not all, NF-κB target genes. And the NF-κB-regulated genes that F14 could not inhibit were the same ones that still recruited BRD4 despite the presence of F14.
Given these observations, we wondered if other acetylated proteins bound to gene promoters served as docking sites for BRD4. To test this idea, we used an inhibitor of BRD4 that prevents the recognition of acetylated proteins. To our surprise, like F14, the inhibitor of BRD4 (a small molecule called JQ1) did not affect the recruitment of BRD4 to some NF-κB-regulated gene promoters. This revealed that the BRD4 recruitment to some genes can be independent of the recognition of acetylated proteins. Therefore, studying a protein from a vaccine of an eradicated disease taught us something fundamental about the regulation of inflammatory genes by NF-κB. In Figure 2, we summarise how F14 inhibits selectively the expression NF-κB target genes.
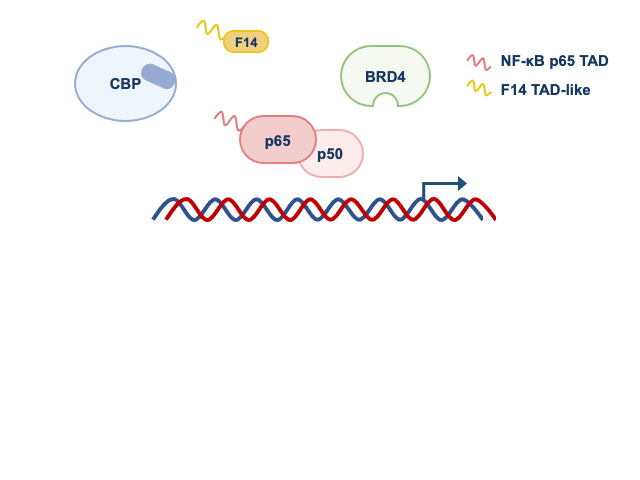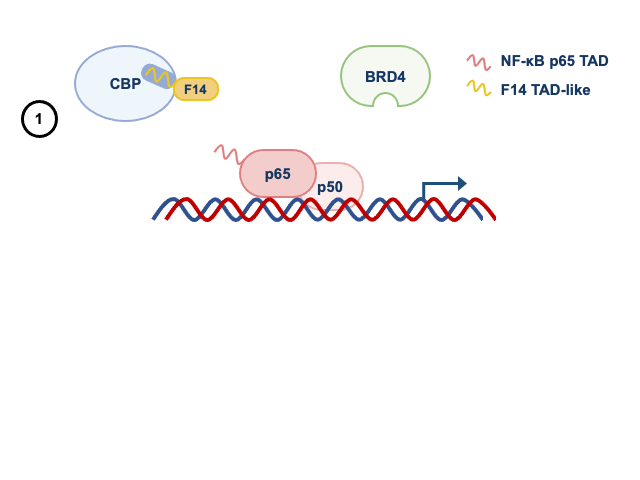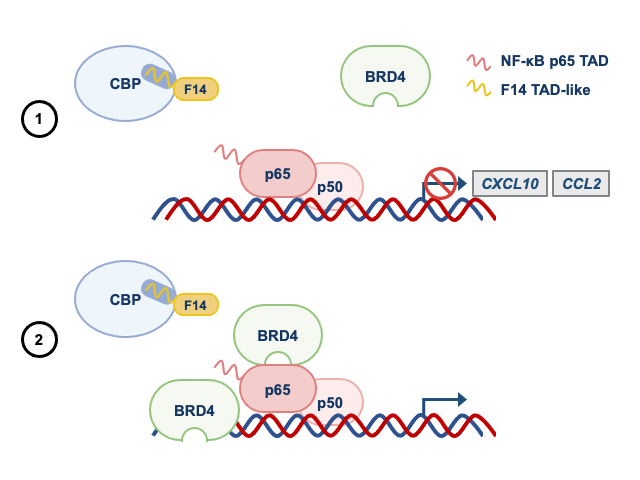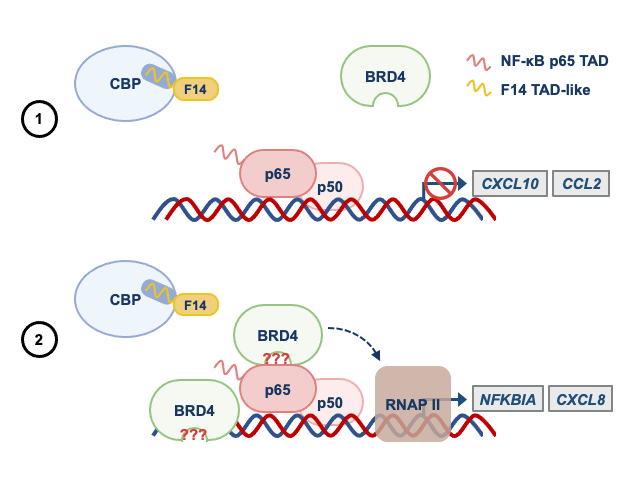
Panel 1 depicts the model for genes that are inhibited by F14, whilst panel 2 depicts that genes that are not inhibited.
See text for more details.
It remains unclear how BRD4 is recruited to gene promoters independently of the recognition of acetylated proteins, but this is likely to be relevant clinically. BRD4 inhibitors are widely sought after for the treatment of a range of human diseases, and normally target BRD4’s ability to recognise acetylated proteins. This knowledge might then help in the development of a new class of BRD4 inhibitors that block its acetylation-dependent and independent functions.
Follow the Topic
-
Nature Microbiology
An online-only monthly journal interested in all aspects of microorganisms, be it their evolution, physiology and cell biology; their interactions with each other, with a host or with an environment; or their societal significance.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in