Introducing MolE: A New Model for Predicting Molecular Properties for AI Drug Design and Beyond
Published in Chemistry, Protocols & Methods, and Computational Sciences
When medicinal chemists are engaged in drug discovery tasks like hit identification, hit-to-lead, and lead optimization, they need to predict properties about the molecules they want to synthesize in order to optimize features like potency and binding affinity while preventing toxicity across a number of different dimensions. The most efficient way to do this is using a trained AI model that can predict the molecule’s biological effects and propose structural changes.
There are a number of these chemical modeling efforts underway, however most of public and private training sets are typically small compared to other fields – in the tens of thousands, versus millions for image recognition labeled examples as with the public image database ImageNet.
Many existing chemical modeling efforts also use a type of molecular representation known as simplified molecular input line-entry system (SMILES) sequences for training. These sequences rely on a linguistic construct, with a series of characters representing the molecules’ atoms and bonds.
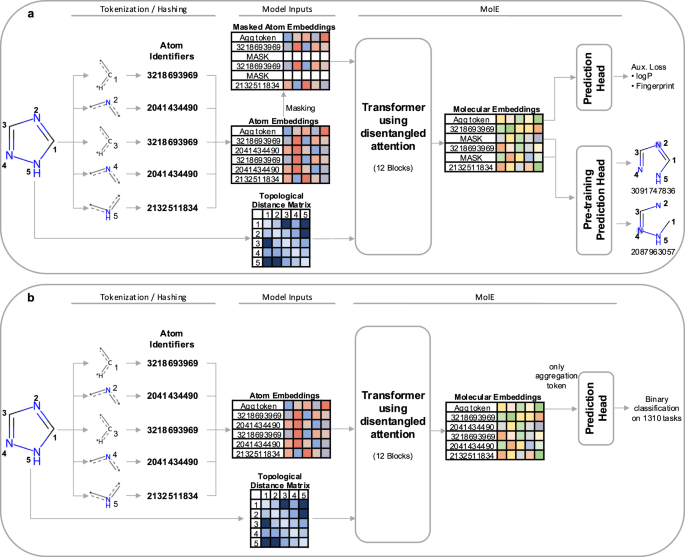
In our paper, we offer a new foundation model for chemistry called MolE that relies instead on molecular graphs – visual depictions of the molecules with nodes and edges as opposed to a linear string of characters. MolE was first trained on over 842 million molecular graphs using a self-supervised approach – meaning it didn’t need experimental results since it learns entirely from the chemical structure – and then further fine-tuned on a set of downstream absorption–distribution–metabolism–excretion–toxicity (ADMET) tasks. Ultimately, we showed that MolE outperforms earlier approaches, ranking first in 10 of the 22 ADMET tasks included in the Therapeutic Data Commons (TDC) leaderboard.
MolE initially learns from unlabeled data, and this allows us to build models with fewer data points, generalizing better and producing better benchmarks in downstream models than prior methods.
Building and Testing the MolE Foundation Model for Chemistry
MolE relies on atom identifiers as input tokens and graph connectivity for positioning information from the molecular graphs. We used this data to pretrain the model – transferring from large unlabeled datasets to smaller labeled datasets by randomly masking atoms and having the model predict the corresponding atom environment of all neighboring atoms separated by no more than 2 bonds. We used both a self-supervised approach with approximately 842 million molecules, followed by supervised pre-training with about 456,000 molecules.

Next, we fine-tuned the model by assessing the quality of its predictions using a set of 22 ADMET tasks included in the TDC benchmark. The Therapeutic Data Commons is a resource providing tools, libraries and other resources accessible via an open Python library, that allows researchers to evaluate AI capability across a variety of therapeutic modalities and stages of discovery. This benchmark gave us a standard way to compare MolE against other established models, such as those using precomputed fingerprints like RDKit or the Morgan fingerprint, convolutional neural networks using SMILES, and versions of graph neural networks like ChemProp.
When compared to the best models in the TDC leaderboard, MolE achieved state-of-the-art performance in 10 of the 22 tasks and was the second-best model on another 4 tasks. The tasks where it performed best included 6 regression tasks and 4 classification tasks, primarily those related to CYP inhibition. CYP enzymes play a major role in metabolizing drugs, and CYP inhibition often leads to drug-drug interactions. While the CYP results are tied to larger datasets, MolE also achieved top performance on tasks with just a few hundred training examples, including predicting half-life and CYP substrates.
The next-best model after MolE, ZairaChem, achieved top performance on only 5 of the 22 tasks.
What We Learned – And What’s Next
This paper demonstrates that we can use a transformer-based model, MolE, to predict chemical and biological properties directly from molecular graphs using a pre-trained model. We used a two-step pre-training approach – self-supervised followed by supervised – to train models that outperformed earlier approaches.
So what could this mean for the future of AI drug discovery?
We think MolE will prove to be an important tool in our understanding of how new molecules will perform in the body – and in guiding the design of highly optimized molecules. By training the model to understand atom environments and their relationship to each other, it can also help avoid problems of classical fingerprints such as sparsity and clashes when using bit vectors.
And while we only used drug-like molecules in this research, we expect that larger and more diverse datasets can only improve the model’s performance. This work, we believe, represents an important first step towards establishing a foundation model for chemical property prediction.
Note to researchers: The code to use the model reported in this study is available under the Attribution-NonCommercial 4.0 International License (CC-BY-NC 4.0) in https://github.com/recursionpharma/mole_public.
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in