Proteases and Philosophy—Decoding Molecular Recognition by Bridging Biochemistry and AI through Explainability
Published in Neuroscience, Protocols & Methods, and Cell & Molecular Biology
“The formulation of a problem is often more essential than its solution […]. To raise new questions, new possibilities, to regard old questions from a new angle requires creative imagination and marks real advance in science."
— Albert Einstein
Why is this research valuable?
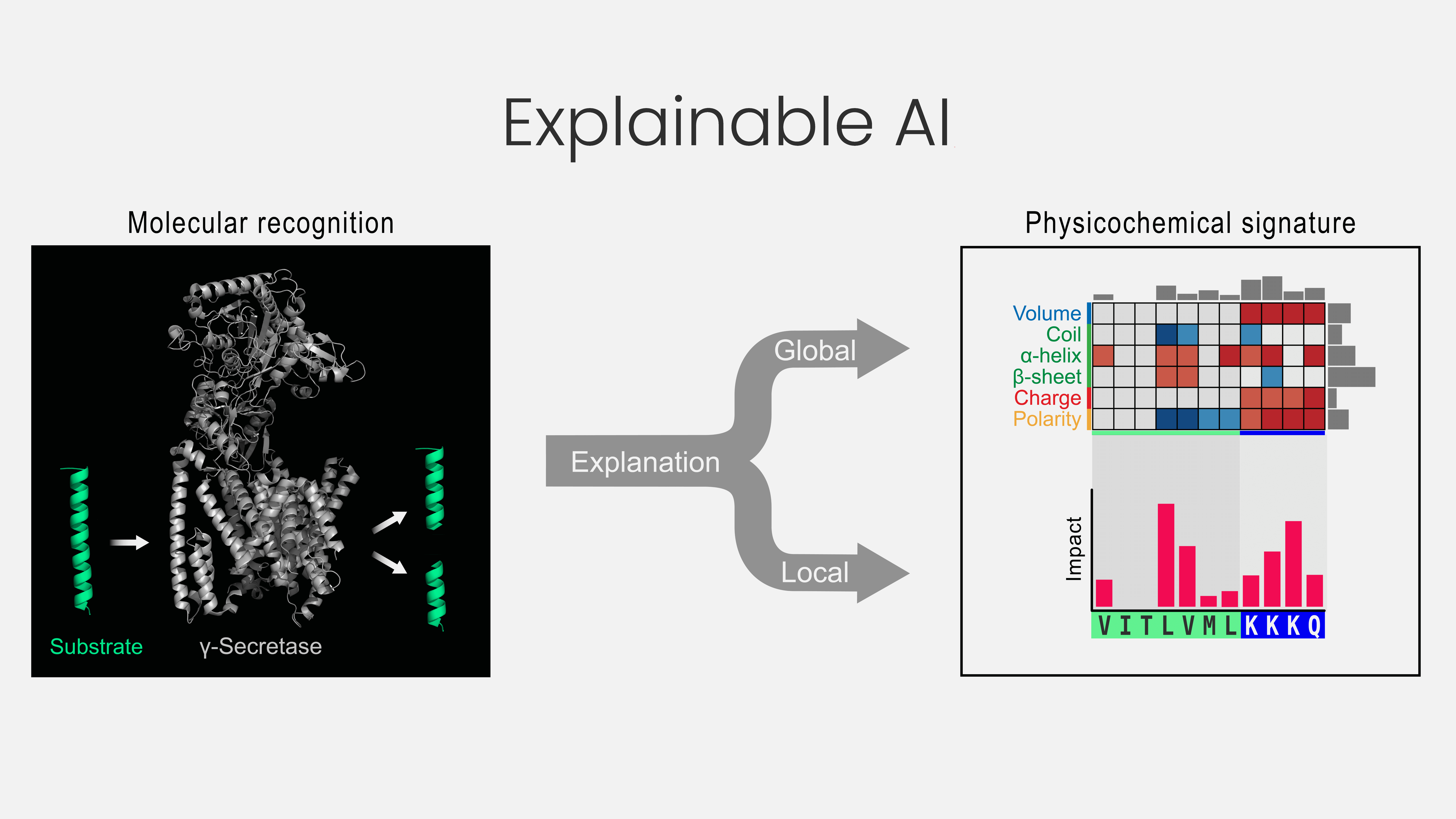
γ-Secretase is a critical enzyme in health and disease, yet how it selects which proteins to cut—its so-called substrates—has remained a mystery, as they lack any recognizable sequence motif. Our new method (CPP) looks beyond sequences, comparing physicochemical properties, such as polarity or charge, across known protein targets. Combined with machine learning and an established explainable AI approach, SHapley Additive exPlanations (SHAP), we not only predict new substrates accurately but also understand why. This fusion of prediction and insight deepens our understanding of how enzymes work and can open new opportunities for protein drug design.
What the authors did
It all began with a simple question: “Is there a hidden, yet unknown property that defines how γ-secretase distinguishes its substrates from non-substrates?” Answering this required more than data. It required crossing disciplinary boundaries.
This journey started during my master’s thesis in molecular biotechnology, supervised by Dmitrij Frishman, a renowned expert in computational protein biology. I attended my first meeting of the intramembrane proteolysis consortium—a vibrant mix of about 30 scientists, from students to institute directors. One discussion remains vivid: Harald Steiner, a leading expert on γ-secretase, illustrated potential substrate recognition mechanisms with sweeping gestures. Dieter Langosch, an expert in biophysics of transmembrane domains (TMD), thoughtfully connected these ideas to what was known about the amyloid precursor protein (APP), the enzyme’s best-known substrate. That moment transformed science for me—from dry textbook knowledge into a process of living discussion creating new knowledge. Later that evening, I sat next to Frits Kamp, a senior scientist and opera singer who would become an intellectual companion throughout this journey.
Meanwhile, I was learning Python and machine learning. I started to wonder: How could we frame this recognition problem computationally? Three key challenges emerged:
- Which part of the sequence defines recognition—just the TMD alone, or the complete sequence?
- TMDs vary in length, making traditional position-wise comparisons ineffective.
- Is being a “substrate” a binary label—or something more gradual?
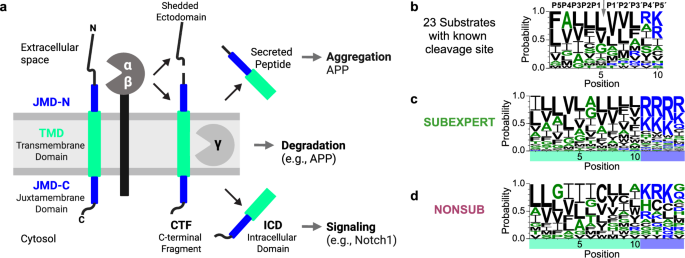
As these puzzles floated in my mind, pieces began to fall into place. At my second consortium meeting, another PhD student presented domain-swapping experiments, showing that the sequence parts adjacent to the TMD, called juxtamembrane domains (JMDs), are also critical for recognition. At the same meeting, Frits and Harald proposed using physicochemical properties to characterize known substrates by profiling them. To achieve this, Dmitrij introduced me to AAindex, a database of such physicochemical property scales. That name inspired the titles of several tools we later developed, including AAontology, AAclust, and AAanalysis. Shortly after, a seemingly trivial discovery while coding—how to split a string in Python—sparked a deeper idea: what if we split protein sequences into meaningful segments and compared those instead of aligning full-length TMDs? This divide-and-conquer logic gave rise to CPP. Yet, one question remained: How do we define a substrate?
While pursuing a second degree in philosophy, I attended a lecture on how European identity was shaped through contrast with non-European cultures. This comparative logic—understanding the essential through contrast—echoes a philosophical approach rooted in Plato and later systematized by Hegel, known as dialectic, which emphasizes thinking in the unity of opposites: thesis (substrate), antithesis (non-substrate), and synthesis (a continuous spectrum between them). I realized: to explain substrates, we must compare them—not in isolation, but against a meaningful reference. Combining this idea with the elements of parts, splits, and scales led to the creation of CPP. Using physicochemical scales, I discovered differences between substrates and a reference set of protein sequences. This was a breakthrough. The initial results, presented during my master´s defense, were promising but puzzling: CPP revealed that substrates have not only an increased helical tendency around the cleavage site but also an unexpected β-sheet propensity at the same positions.
When I shared these findings with Frits and Harald, they weren’t surprised, they were excited. A new cryo-EM structure had just shown that the cleavage region, although helical in the unbound state, unfolds and forms a hybrid β-sheet with γ-secretase upon binding. This explained why substrate sequences must possess the potential to switch between two structural states, a transition triggered by the substrate–enzyme interaction. The biological insight mirrored a philosophical one. Wittgenstein’s “language games” suggest that meaning depends on context. The same, I realized, applies to proteins: function emerges from a sequence’s ability to adopt multiple conformations. After this meeting with Harald and Frits, Dieter shared with me a quote by Thomas Edison: “Innovation is 1% inspiration and 99% perspiration.” He was right about the work that lay ahead of me.
The spark—first lit in the lively discussion between Harald and Dieter—grew into a fire, fueled by a defining moment in Heidelberg. During my presentation, Christian Haass, a leading Alzheimer’s researcher, stood up and said: “We’ve done it. We’re finally on the right track to solve this long-standing question.” That moment of recognition kept me going through every setback that followed.
Then the COVID-19 lockdown hit. Isolated but focused, I entered my most productive coding phase. A close friend introduced me to a new explainable AI method, SHAP. One image crystallized in my mind: a CPP profile with SHAP values showing how each single residue contributes to recognition. That vision stayed with me; it emerged first as an image and then slowly took form in code over three intense months. From that effort, the CPP‑SHAP profile was born.
After COVID-19 restrictions lifted, I faced another core challenge: imbalanced data. We had many substrates, but few experimentally confirmed non-substrates. Simulated negatives felt biologically unsound—proteins are sensitive, and even small changes can flip function. At a summer school, I heard about Positive-Unlabeled (PU) learning. I explored the literature but couldn’t find reproducible solutions. Weeks of frustration followed. Then, one night, I woke up with the idea fully formed. I got out of bed and coded until morning. The result: a deterministic PU learning algorithm (dPULearn). It proved to be the missing piece.
The following years were filled with iterations: method refinement with Dmitrij, even during his vacations (I still remember calls from a poolside in Tuscany and a snowbound hotel in the French Alps); working on visual storytelling with Frits at Café Mariandl, learning that science is not just precision—it’s communication until understanding is shared; rounds of manual dataset curation with Frits and Harald; and experimental validation, a tour de force by Gabi and Claudia, Harald’s exceptional technical assistants. Finally, countless hours went into crafting and wordsmithing the manuscript with Harald and Frits, chasing clarity until every sentence reflected a common understanding.
This paper is the result of that shared intellectual journey. To me, science is both an art and a craft—a process of thinking together across disciplines, generations, and perspectives. Or, as Frits says, adapting a thought of Hannah Arendt: “Science is not about revealing an everlasting truth, but about being truthful and providing reliable predictions.”
Conclusion
The story behind this paper is not just γ-secretase—it’s about scientific creation. Over this journey, I experienced three distinct forms of it:
- Creativity: A string-splitting function, philosophical ideas, and scientific debates shaped CPP.
- Vision: A vivid mental image turned after months of intense work into the CPP-SHAP profile.
- Suffering: Weeks of frustration over an imbalanced data problem gave rise to dPULearn.
Our work reflects what science is at its core: a shared search for understanding across disciplines and generations. It’s not just about solving problems, but asking the right questions, bridging ideas, and building tools others can use. Science is art in conception, craft in execution, and meaning in communication—above all, a journey made richer by those who walk it together.
What are the implications of this study?
- For Alzheimer’s and γ-secretase biology: We provide an answer to a long-standing—perhaps partly forgotten—question: how γ-secretase recognizes its substrates. Our findings reveal a broader and more diverse substrate spectrum than previously anticipated, including immune- and cancer-related proteins validated experimentally. Many substrates act as functional hubs, suggesting broader roles for γ-secretase in health and disease.
- For computational biology and explainable AI: Our alignment-free, interpretable framework performs well even on small and imbalanced datasets, conditions common in biological research. By combining domain knowledge with CPP- and SHAP-based explainability, we show that machine learning can yield mechanistic insight, not just predictions—turning explainable AI into a discovery engine.
- For conceptual thinking in protease biology: Many proteases are promiscuous enzymes. Rather than relying on fixed sequence motifs or structural snapshots, we extract physicochemical patterns capturing dynamic, context-dependent behavior—properties directly encoded in the sequence. This reframes substrate definition as a complex signature rather than a fixed rule, an approach with potential to decode other molecular recognition processes.
Looking ahead
We aim to generalize this framework into a mechanism-aware platform for explainable protein design—one that predicts function and reveals the logic behind it, bridging AI and biology.
Related material
For readers interested in the methods and tools behind this journey:
- Breimann & Kamp et al. (2025), Charting γ-secretase substrates by explainable AI, Nature Communications
- Breimann & Frishman (2024), AAclust: k-optimized clustering for selecting redundancy-reduced sets of amino acid scales, Bioinformatics Advances
- Breimann et al. (2024), AAontology: An ontology of amino acid scales for interpretable machine learning, Journal of Molecular Biology
- AAanalysis (Python framework)
Follow the Topic
-
Nature Communications
An open access, multidisciplinary journal dedicated to publishing high-quality research in all areas of the biological, health, physical, chemical and Earth sciences.

Related Collections
With Collections, you can get published faster and increase your visibility.
Women's Health
Publishing Model: Hybrid
Deadline: Ongoing
Tumor Microenvironment Crosstalk and Therapeutic Implications
Publishing Model: Hybrid
Deadline: Nov 02, 2026




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in